Traduzione di Natale
Marzari
Traduzione di Natale
Marzari
Home,
Ricerca,
Indice alfabetico,
Collegamenti,
Patologia,
Molecole,
Sindromi,
Muscoli,
Giunzioni neuromuscolari,
Nervi,
Spinale,
Atassia,
Anticorpi e Biopsia,
Informazioni per i
pazienti
|
 |
Traduzione del 23 settembre 2010
MIOPATIE E
DEBOLEZZE CONGENITE
|
DEBOLEZZE CONGENITE |
Assenza dei muscoli
Actina, aggregati di: ACTA1; 1q42
Apoptosi
Artrogriposi (Piede a clava)
Autofagia eccessiva: VMA21; Xq28
Bethlem, miopatia
COL6A1: 21q22
COL6A2: 21q22
COL6A3: 2q37
Banda A larga
Bastoncelli nemalinici
Actina α: 1q42
TPM3: 1q21
TPM2: 9p13
CFL2: 14q12
Nebulina: 2q21
RYR1: 19q13
TNNT1: 19q13
Lento movimenti: 15q21
Campodattilia Tel Hashomer
Cap: TPM2; 9p13
Carey-Fineman-Ziter, Sindrome
Centronucleare (Miotubulare)
DNM2: 19p13
RYR1: 19q13
MYF6: 12q21
BIN1: 2q14
MTM1: Xq27
Corpi a impronta digitale
Corpi citoplasmatici (Sferoidali)
Corpi di Mallory: SEPN1; 1p35
Corpi ialinici (miosina accumulo): MYH7; 14q11
Corpi riducenti: FHL1; Xq26
Corpi sferoidali: MYOT; 5q31
Corpi zebrati: Actina α; 1q42
Danon: LAMP2; Xq24
Diarrea e sordità
|
Sproporzione dimensionale fra tipi di fibre
Actina α: 1q42
SEPN1: 1p35
CFTD2: Xq13
TPM3: 1q21
Distonia rispondente alla DOPA
Distrofia FSH: 4q35
Distrofia miotonica 1: DMPK; 19q13
Distrofie muscolari congenite
Disturbi metabolici mitocondriali
Disturbi scheletrici + Debolezza
Fibre di tipo 1 piccole
Filamenti sottili, Eccesso: Actina α; 1q42
Focale
IBM3: MYH2; 17p13
Miasteniche, sindromi
Miopatia dei nativi americani: 12q13
Miopatia letale congenita: CNTN1; 12q11
Miopatia perifascicolare, neonatale
Miopatia trilaminare
Multinuclei (Mininuclei)
RYR1: 19q13
SEPN1: 1p35
Neuropatiche, sindromi
Nucleo centrale, malattia del: RYR1; 19q13
Oftalmoplegie
Ritardo mentale, collegato a X: Xq24
Sarcotubulare (LGMD
2H): TRIM32; 9q31
Williams-Beuren, sindrome
Woods-Black-Norbury sindrome |
|
DISTROFIE MUSCOLARI CONGENITE
Atrofia cerebellare
Atrofia del CNS e assenza grandi assoni PN mielinizzati
CMD: 4p16
CMD + Cardiomiopatia: Titina; 2q24
Desmina inclusioni: SEPN1; 1p36
Disturbi
ipertrofia muscolare
Ritardo mentale
Insufficienza respiratoria (MDC1B): 1q42
ipertrofia muscolare (MDC1C): FKRP; 19q13
Grave ritardo (MDC1D): LARGE; 22q12
Spina rigida con CMD
SEPN1: 1p35
Lamina A/C: 1q21
Altre sindromi della spina rigida
Epidermolisi bollosa giunzionale: Plectina; 8q24
Fukuyama: Fukutina; 9q31
Integrina α-7: 12q13
Iper rilassatezza delle articolazioni: 3p23
Marinesco-Sjögren: SIL1; 5q31
Merosina (laminina α2-catena)
Carente (MDC1A): 6q22
Normale: "Pure" forma
Mitocondriali alterazioni strutturali
muscoli-occhi-cervello
Pollici addotti e oftalmoplegia
Santavuori (Finnica): POMGnT1; 1p32
Ullrich
COL6A1; 21q22
COL6A2; 21q22
COL6A3; 2q37
Walker-Warburg
FKRP: 19q13
Fukutina: 9q31
LARGE: 22q12
POMGnT1: 1p32
POMT1: 9q34
POMT2: 14q24
Caratteristiche generali
Altre debolezza congenita
|
ASSENZA O DEBOLEZZA DEI
MUSCOLI
Addominale
Plesso-brachiale
Sindrome cardiofacciale
Depressore anguli oris
Diaframma
Muscoli extraoculari
Blefarofimosi
Fibrosi congenita
Duane, sindrome di
Möbius, sindrome di
Ptosi
Retto superiore
Eminenza tenar
Estensori delle dita
Holt-Oram
Palmaris longis
Pettorale
Peroneus terzo
Poland, sindrome di
Prune belly
Psoas (CHILD)
Trapezio
Batten
|
GENERALE
- Ipotonia: Valutazione clinica
- Risposta alla trazione
- Manovra: Prendere le mani del bambino; Tirare fino alla posizione seduta
- Neonati: Il capo rimane all'indietro; Cade improvvisamente in avanti improvvisamente quando viene raggiunta la posizione eretta
- Normale a 1 mese: Minima sfasamento del capo; Flessione dei gomiti,
delle ginocchia,
e delle caviglie
- Infanti prematuri < 33 settimane di gestazione: Assenti
- Sospensione: Verticale e orizzontale
- Valutazione della madre
- Storia: Ritardo nel camminare o altre funzioni motorie; Deformità dello scheletro o delle articolazioni
- Forza
- Miotonia
- La malattia del CNS è la causa più comune della ipotonia congenita
- Caratteristiche comuni di presentazione del disturbo neuromuscolare
negli infanti
- Ipotonia
- Postura con tono normale: Flessione di ginocchia, anche, e gomiti; Anche
ruotate verso l'interno
- Posture con ridotta tone: Capo ricadente; Gambe e braccia estese
- Debolezza: Livelli simili nei muscoli facciali, appendicolari, e assiali
- Massa muscolare: Ridotta
- Sensibilità normale
- Disturbi scheletrici
- Le meglio definite cause neuromuscolari di debolezza congenita
includono
- Diagnosi di sindromi con debolezza congenita
- Spesso basata su risultanze morfologiche della
biopsia muscolare
- Sindromi di debolezza congenita distinte clinicamente
l
Recettore rianodina (RYR1; RyR)
 ; Cromosoma 19q13.1;
Dominante o recessiva
; Cromosoma 19q13.1;
Dominante o recessiva
- Caratteristiche genetiche 11
- Identificate 251 mutazioni
- Tipi di mutazione: La maggior parte missenso; Qualche piccola delezione; Rare
splice o inserzione
- La maggior parte delle famiglie nucleo centrale collegate a mutazioni del recettore rianodina
- Alcune mutazioni puntiformi producono solo la MH: La maggior parte nella
regione del terminale N
- Arg614Cys: Comune (4% ÷ 9%)
- Produce anche la MH suina
- Soglia più alta e contratture più piccole comparata ad altre mutazioni
- Gly2433Arg: Comune (4% ÷ 7%)
- Gly341Arg: 5% ÷ 6% delle famiglie britanniche ed irlandesi
- Arg2454Cys: Bassa soglia scatenante
- Altre: Gly248Arg; Arg552Trp; Arg614Leu; Val2168Met; Thr2206Met
- Alcune mutazioni con malattia del nucleo centrale e MH
- Arg163Cys; Ile403Met; Tyr522Ser; Arg2434His; Arg2454His
- Fenotipo grave e penetrante: Ile4898Thr
- Genealogia messicana
- Regioni transmembrana/luminali del terminale C
- Proprietà del canale
- Permeabile
- Attivato da concentrazioni del Ca++ 4 volte più bassa del normale
- Mutazione del recettore rianodina con malattia del nucleo centrale,
bastoncelli e MH: Y4796C
- Punti caldi della mutazione
- Regione citosolica della proteina
- Terminale N citoplasmatico: Mutazioni fenotipiche delle ipertermia maligna in questa regione
- Regione citoplasmatica centrale: Segmento genetico 6400-6700
- Le mutazioni spesso avvengono nella sequenza dinucleotide CpG del filamento codificante o antisenso del DNA
- 3' dominio transmembrana (esoni 93-105): 30% dei casi
- Ipertermia maligna all'IVCT
- Ma nessuna complicazione clinica con l'esposizione ad agenti scatenanti la MH
- Mutazioni: Altre
- 2 differenti mutazioni nei codoni 614 (lo stesso della 615 suina), 2163, 2454 e
2458
- 1 Mutazione nel terminale C: Fenotipo grave
- Proteina recettore rianodina 81
- 560 kDa
- Struttura
- Tetramerica
- 6 ÷ 8 domini transmembrana
- Il 20% delle molecole del terminale C formano il canale
Ca++ transmembrana
- Terminale N: Contiene la maggior parte
dei siti di legame del ligando; Dominio citoplasmatico
- Molecole regolatorie
- Ca++
- Mg++: Inibitorio
- Nucleotide adenina e ADPribosio ciclico: Attivazione del canale
- Ossidazione
- Nitrosilazione
- Caffeina e 4-chloro-m-cresol (CmC)
- Attivano i canali RyR
- Facilitano l'apertura del canale: Aumentando la sensibilità dei RyR all'attivazione dipendente
dal Ca++
- Bloccanti dei RyR; Rutenio rosso; Anestetici locali (Procaina; Tetracaina)
- Dantrolene: Inibisce la permeabilità Ca++ del SR via RyR1
mutante; Usata per
prevenire la crisi MH
- Proteine associate
- Recettore diidropteridina
- Proteina legante FK506
- Calmodulina (CaM): Partecipa alla regolazione dell'attività del canale
dipendente dal Ca++
- Ca++ non legato a CaM: Attiva RyR
- Ca++ legato a CaM: Inibisce la funzione RyR del canale
- Triadina
- Calsequestrina
- Chinasi: PKA; CamK
- Fosfatasi: PP1, PP2A
- Fosfodiesterasi PDE4D3
- Proteina chinasi A di ancoraggio muscolare
- Isoforme
- RYR1: Muscoli scheletrici
- RYR2
: Cardiaco
- RYR3
: CNS
- Funzioni
- Strutturali: Il piede della struttura colma la lacuna (gap) tra il
reticolo sarcoplasmatico e
i tubuli t nei muscoli scheletrici
- Accoppiamento eccitazione-contrazione
- Aumenta il Ca++ necessario per la contrazione delle fibre muscolari
- Canale di rilascio del Ca++
dei muscoli scheletrici
- Provvede al rilascio del Ca++ dal reticolo sarcoplasmatico
con l'attivazione dei recettori rianodina nei canali
- Il Ca++ rilasciato dal reticolo sarcoplasmatico si accumula (lumen) dentro
il
citoplasma
- Funzionalmente accoppiato a variazioni del potenziale di membrana sarcolemmale
attraverso i VGCC di tipo L (DHPR)
- Apertura dei RyR1: Indotta da cambiamenti conformazionali nei DHPR a
seguito della
depolarizzazione di membrana (Interazione fisica)
- L'attivazione dei RyR2 nel cuore: L'attivazione dei canali
Ca++ di tipo L induce il rilascio di Ca++ indotto da Ca++ (CICR)
- Effetti della mutazione sulla funzione della proteina: Più MHS e CCD con
- Maggiore sensibilità della proteina RYR mutante agli agonisti caffeina e
alotano
- Più alto il Ca++ nel sarcoplasma a riposo
- Minore accumulo del Ca++ nel reticolo endoplasmatico
- Ridotta quantità del Ca++ accumulato rilasciabile
- Y522S: RyR1 permeabile
- Terminale C (14895T e altro): Disgiunge l'accoppiamento EC
- Caratteristiche cliniche
- Insorgenza: Insorgenza congenita o nella fanciullezza
- Genetiche
- Punto caldo della mutazione: Terminale C esoni 101–102
- Sviluppo
- Movimenti fetali ridotti
- Presentazione podalica
- Motorio
- Ipotonia
- Debolezza prossimale: Gambe > braccia
- Faccia: Debolezza lieve
- Movimenti extraoculari: Normali
- Crampi
- Scheletro: Lussazione congenita dell'anca; Scoliosi; Piedi deformità
- Decorso: Non o lentamente progressiva
- Varianti: Sindrome del nucleo centrale sindromi e altri disturbi RyR
- Ipertermia maligna: Insorgenza nell'adolescenza
- Sindrome King-Denborough
- Sindrome dei cingoli degli arti: Insorgenza nell'adolescenza
- CK serica alta: Asintomatica
- Mutazione del recettore rianodina con malattia del nucleo centrale, Bastoncelli
e Ipertermia maligna
- Mutazioni
- Y4796C: Dominio di formazione del terminale C del canale
- Thr4637Ala: Regione transmembrana
- Epidemiologia: 2 famiglie
- Clinica
- Insorgenza: Infantile
- Debolezza: Non progressiva; Ipotonia; Camminata a 2 anni e mezzo
- Contratture: Caviglie
- Decorso: Rimane la deambulazione nel 3° e 4° decennio
- Laboratorio
- CK serica: Normale
- Biopsia muscolare: Nucleo centrale; Bastoncelli in aree senza nuclei
- Esame di contrattura in vitro: Positivo
- Altre sindromi bastoncelli-nucleo: Mutazioni Nebulina
- Malattia del nucleo centrale con
mininuclei transienti
30
- Genetica
- Ereditarietà: Recessiva
- Mutazione missenso omozigotica (P3527S)
- Epidemiologia: Famiglie algerine, turche e tedesche
- Clinica
- Insorgenza: Infanzia; Ipotonia
- Debolezza
- Gravità
- Solitamente moderata
- Variabili all'interno delle famiglie: Alcuni pazienti con grave disabilità
- Distribuzione
- Muscoli assiali: Collo; Flessori del tronco
- Cingoli pelvici
- Mani: Debolezza moderata; Atrofia
- Faccia: Alcuni pazienti
- Decorso: Stabile o lentamente progressivo
- Articolazioni
- Iperrilassatezza: Coinvolgimento delle mani; Coinvolte anche altre articolazioni;
Lussazione del ginocchio e della rotula
- Contratture: Alcuni pazienti; Lievi; Principalmente dei calcagni
- Laboratorio
- CK serica: Normale
- Cardiaco: Normale
- MRI: Preferenzialmente coinvolti gluteo massimo e quadricipite
- Biopsia muscolare
- Fanciullezza: Mininuclei multipli
- Adulti: Nucleo centrale; Perdita di fibre muscolari
- Predominanza delle fibre muscolari di tipo 1
- Sindrome con malattia del nucleo centrale o multinuclei
- Genetica: Mutazione missenso Arg 4893Trp; Eterozigotica
- Ereditarietà dominante
- Malattia del nucleo centrale con acinesia fetale
45
- Ereditarietà: Dominante o recessiva
- Mutazioni
- Recessive
- R614C (Spesso associata con MH) e G215E
- L4650P e K4724Q
- Dominante: G4899E (Madre colpita meno gravemente)
- Clinica
- Gravidanza
- Postnatale
- Ipotonia: Grave
- Sofferenza respiratoria
- Malformazioni multiple
- Decorso
- Morte per alcuni
- Altri con debolezza stabile dopo prolungato supporto respiratorio
- Oculare: Possono svilupparsi strabismo e ptosi
- Patologia muscolare
- Nuclei centrali: Eccentrici
- Tessuto connettivo endomisiale: Aumentato
- Malattia del nucleo centrale: Fenotipo lieve ed ereditarietà recessiva
- Genetica
RYR1
- Mutazione missenso V4849I
- Stessa mutazione trovata anche nella famiglia della ipertermia maligna dominante
- Malattia modello sovrapposto al fenotipo lieve della
malattia multinuclei
- Malattia neuromuscolare congenita con fibre di tipo 1 uniformi (CNMDU1)69
- Genetica RYR1: Mutazioni
- Eterozigotiche
- Missenso o delezione
- Localizzazione: Terminale C del dominio di RYR1; Poro formante segmento
- Mutazioni trovate nel 40% dei pazienti con CNMDU1
- Nessuna nei pazienti con ritardo mentale
- Clinica: Miopatia congenita
- Insorgenza: Congenita o nella prima infanzia
- Debolezza: Lieve; Prossimale; Faccia normale
- Riflessi tendinei: Ridotti o assenti
- CNS: Nessun ritardo mentale
- Laboratorio
- CK serica: Normale
- EMG: Miopatica
- Biopsia muscolare
- Tutte fibre muscolari di tipo 1 (> 99%)
- Nessun nucleo
- Tessuto connettivo endomisiale: Normale o lieve aumento
- Anticorpi contro RyR
- Mutazioni RyR2
- Laboratorio
- CK
- Alta nei pazienti con nucleo centrale e MH
- La CK può essere alta in alcuni portatori asintomatici (G341R)
- Esame di contrattura in vitro per ipertermia maligna
- Sensibilità: 97% ÷ 99%
- Specificità: 78% ÷ 94%
- MRI muscolare 52
- Caratteristiche del coinvolgimento muscolare
- Segnali muscolari: T1 aumentato
- Correlazione clinica: Modelli simili con differenti sindromi cliniche e
mutazioni RYR1
- Coscie
- Coinvolti: Vasti, sartorio, adduttore magno
- Risparmiati: Retto, gracile, adduttore lungo
- Basse gambe
- Coinvolti: Soleo, gastrocnemio, peronei
- Risparmiati: Tibiale anteriore
- Malattia del nucleo centrale: Patologia muscolare
- Miopatia
- Dimensioni variabili delle fibre muscolari
- Aumento del tessuto connettivo endomisiale
- Nuclei interni: Specialmente nei pazienti più anziani
- Nuclei: Zone interne, "nuclei", nelle fibre muscolari
- Morfologia dei dischi Z alterata
- Ridotti o assenti nei nuclei
- Attività enzimatica ossidativa
- Mitocondri
- Alcuni nuclei hanno anche perdita della struttura miofibrillare centrale
- I nuclei centrali corrono per l'intera lunghezza delle fibre muscolari
- Più evidenti nelle colorazioni NADH e mitocondriale
- Immunoreattività nei nuclei 36
- Proteine miofibrillari
- Cristallina αB; Desmina; Filamina C
- Modelli simili di immunoreattività si vedono nei bersagli
- Proteine correlate al Ca++
- Depleta dai nuclei: RyR1
- Accumulate nelle, o attorno alle lesioni
- Altre proteine del reticolo sarcoplasmatico: Calsequestrina, SERCA1/2 e
triadina
- Proteine dei tubuli t: Subunità α1 del recettore diidropiridina
- Mutazione SelN correlata ai mininuclei: Normale distribuzione delle
proteine correlate al Ca++
- Marcata predominanza delle fibre muscolari di tipo I
- Correlazione clinica
- Nuclei ben formati + marcata predominanza del tipo 1
- Sindrome miopatica: Spesso senza MHS
- Mutazioni RYR1: 90% con 60% nel terminale C
- Nuclei variabili + minore predominanza del tipo 1 + nuclei interni
- Sindrome MHS: Solitamente forza normale
- Mutazioni RYR1: 35%, all'esterno del terminale C
Gestione: Evitare l'ipertermia maligna correlata all'anestesia
l
Miosina - Catena spessa β cardiaca (MYH7)
; Cromosoma 14q11.2-q13;
Dominante
- Caratteristiche cliniche
- Biopsia muscolare
- Nuclei centrali e predominanza delle fibre muscolari di tipo I
- Altre cardiomiopatie ipertrofiche familiari hanno una normale istologia dei
muscoli scheletrici
Miopatie nemaliniche (Bastoncelli)
Miopatie con bastoncelli: Caratteristiche cliniche generali 22
- Insorgenza
- Ereditarietà: Dominante, recessiva e sporadica
- Mutazione comune: Muscoli scheletrici actina α
- Casi ad insorgenza precoce: Comuni insufficienza respiratoria e difficoltà di alimentazione
- Caratteristiche cliniche
- Motorio: Debolezza e ipotonia
- CNS: Coinvolgimento cognitivo nei pazienti con insorgenza più giovane
- Cardiaco: Raramente sintomatico dopo il periodo neonatale
- Artrogriposi: Grave nei casi sia congenita
- Paraspinoso o
debolezza posteriore del collo: Nei pazienti ad insorgenza adulta
- Decorso
- Morbilità alle infezioni respiratorie e problemi di alimentazione minori con
l'aumentare dell'età
- Camminare
- Raro nella miopatia a bastoncelli congenita
- Nella insorgenza infantile solitamente rimane la capacità di ambulare
- Mortalità
- Morti dovute a insufficienza respiratoria
- Precoce aumenta con: Insufficienza respiratoria ; Artrogriposi; Incapacità
a conseguire le prime pietre miliari motorie
- Caratteristiche variabili dei singoli geni
Miopatie con bastoncelli: Caratteristiche generali di laboratorio
- Proteine mutanti: Solitamente componenti strutturali dei filamenti sottili
- Patologia muscolare: Disturbi dei filamenti sottili
- Bastoncelli
- Apparenza istochimica: Strutture rosso-blu scure visibili solamente alla colorazione tricromica di Gomori
- Distribuzione fra i tipi di fibre: Tipo I > II
- Dimensione: 1 ÷ 7 μm di lunghezza
- Composizione: Contengono actinina α + actina + altre proteine della
linea Z
- Ultrastruttura
- Dense di elettroni
- A forma di bastoncelli
- Emanate dalle linee Z
- Inspessanti le linee Z, o
- Estesi lungo l'asse dei filamenti sottili
- Localizzazione subcellulare: Originati dalle
linee Z
- Miopatia: Tendenti a raggrupparsi sotto il sarcolemma e attorno ai nuclei
- Fibre bersaglio: Centro del bersaglio
- Processo patogeno sottostante
- Formazione dei bastoncelli 2° alla disfunzione contrattile
- Probabilmente coinvolti i processi dipendenti dal carico
- Spesso aumentano di numero con età
- Diagnosi differenziale
- Fibre muscolari di tipo I: Sottili; Predominanti
- CK: Normale (90%) o ± elevata nelle forme ad insorgenza adulta
- EMG: Miopatica o neuropatica (Specialmente casi gravi)
- Le mutazioni più comuni: Nebulina;
ACTA1
Miopatie con bastoncelli: Sindromi specifiche
:
l
NEM1 Miopatia con bastoncelli
:
Tropomiosina α 3 (TPM3)
Cromosoma 1q21-q23; Dominante o recessiva
- Proteina TPM3
- Muscoli: Espressa nelle fibre muscolari di tipo I
- 11 forme differenti: Espresse in molti tessuti compreso il cervello
- Vedi: Proteine delle fibre muscolari
- Correlazioni genetico-cliniche
- Infrequente causa di miopatia nemalinica con bastoncelli
- Frequenza: < 3%
- Sindrome più lieve: Mutazione missenso; Ereditarietà dominante
- Grave sindrome: Codone di stop; Ereditarietà recessiva
- Ereditarietà dominante
- Mutazioni: Met9Arg; Arg167His 38
- Insorgenza: 0 ÷ 15 anni
- Clinica
- Debolezza
- Sviluppo: Ritardo motorio in alcuni pazienti
- Distribuzione
- Simmetrica
- Gambe distale (dorsiflessori delle caviglie)
- Braccia
- Insufficienza respiratoria
- Debolezza prossimale: Successivamente nel decorso della malattia
- Progressione: Lenta; Carrozzina spesso a 40 anni
- Riflessi tendinei: Ridotti
- Acalasia: Coinvolgimento della muscolatura liscia
- Morfologia
- Ridotta
- Fisico magro
- Faccia lunga
- Laboratorio
- CK serica: Normale
- Patologia
- Fibre di tipo I: Atrofia; Predominanza
- Bastoncelli: Nelle fibre di tipo I; Può essere infrequente
- Ereditarietà recessiva
- Insorgenza precoce, malattia grave
- Mutazione: Omozigotica; Stop al codone 31
- Insorgenza: Nascita
- Sviluppo motorio: Estremamente ritardato e insufficiente
- Cognitivo: Appropriato all'età
- Morte: 21 mesi
- Patologia
- Bastoncelli: Solo nelle fibre di tipo 1
- Fibre muscolari di tipo 1 assottigliate
- Perdita di tropomiosina α 3 dai muscoli
- Sindrome intermedia 35
- Storia familiare: Sporadica
- Genetica
- Composizione eterozigotica
- Mutazione: X285Ser; Sito di splicing al segnale traslazionale di stop
specifico dei muscoli scheletrici
- Proteina mutata: contiene altri 57 aminoacidi addizionali
- Clinica
- Insorgenza: Nascita; Ipotonia
- Motorio
- Camminata: 18 mesi
- Carrozzina: 6 anni
- Debolezza: Prossimale
- Faccia: Lunga e stretta; Palato ad arco alto
- Patologia muscolare
- Bastoncelli: Nelle fibre muscolari di tipo I
- Tessuto connettivo endomisiale: Aumentato
- Predominanza di fibre muscolari di tipo I
- Nuclei interni
- Fibre di entrambi i tipi sottili
- Ridotta tropomiosina β nelle fibre muscolari
- Sindrome variante: Sproporzione congenita fra i tipi di fibre con mutazioni TPM3
70
l Dominante o sporadica
- Generale
- La più comune causa di CFTD: ~20% ÷ 25%
- Fibre di tipo 1: Almeno il 50% più sottili di quelle di 2
- Fibre di tipo 2: Normali o di grandi dimensioni
- Genetiche
- Mutazioni: Missenso; Eterozigotiche
- Localizzazioni: Leu100Met, Arg168Cys, Arg168Gly, Arg168His, Lys169Glu,
Arg245Gly
- La mutazione Arg168Cys: Può essere associata con CFTD ed alla patologia
della miopatia nemalinica nella stessa famiglia
- Clinica
- Insorgenza
- Età
- Solitamente < 1 anno
- Variabile: Persino dentro la famiglia
- Ipotonia
- Scarso controllo del capo
- Debolezza
- Prossimale > distale
- Gambe > braccia
- Collo: Flessione ed estensione
- Caviglia: Dorsiflessione
- Tronco: Paraspinale; Addominale
- Facciale: Lieve
- Ptosi: Lieve; Unilaterale o bilaterale
- Respiratoria
- Frequenza: 90%
- Ventilazione notturna non invasiva : Insorgenza 3
÷ 55 anni
- Persino in alcuni pazienti deambulanti
- Scapole alate: Lieve; 40%
- Quadricipiti:
Relativamente preservati
- Decorso
- Miglioramento in forza e funzione con il tempo
- I pazienti spesso hanno la possibilità di camminare o correre
- Altri muscoli
- Amiotrofia: Generalizzata
- Struttura corporea: Sottile
- Andatura: Anserina; Piedi ricadenti
- Scheletro
- Bambini giovani: Positura ipotonica; Lordosi lombare e cifosi toracica
esagerate
- Tarda infanzia o età adulta: Cifoscoliosi da lieve a grave con contratture
degli estensori del collo
- Altre contratture: Insolito
- Laboratorio
- EMG: Normale o miopatica
- NCV: Normale
- Cardiaco: Normale
- Patologia muscolare
- Fibre muscolari di tipo 1
- Dimensione ridotta
- 60% del normale; Gamma 30% ÷ 80%
- 23% ÷ 50% delle dimensioni del tipo 2
- Predominanza: Minoranza dei pazienti
- Fibre muscolari di tipo 2
- Dimensione: Grandi; 160% del normale; Gamma 110%
÷ 220%
- Tipo 2B: Assenti
- Tipo 2C: Nessuna o qualche una
- Nuclei interni: Nei pazienti più anziani; Fino a 25% delle fibre di tipo 2
- Nessun bastoncello
- Vedi anche: Miopatia con bastoncelli nemalinici
- Sindrome variante: Miopatia Cap con mutazioni TPM3
- Epidemiologia: 1 paziente
- Mutazione TPM3: Arg168Cys
- Clinica
- Pietre miliari motorie ritardate
- Cifoscoliosi,
- Atrofia muscolare: Generalizzata
- Debolezza: Lieve, Camminata senza supporti; Respiratoria
- Dismorfismi: Faccia allungata; Palato ad arco alto
- Laboratorio
- Biopsia muscolare: Cap, NADH+
l
Miopatia con bastoncelli NEM2:
Nebulina
Cromosoma 2q21.2-q22; Recessiva
- Epidemiologia
- La forma più comune della "tipica" miopatia nemalinica con bastoncelli
recessiva
- Mutazioni
- Trovate 64 mutazioni in 55 famiglie
- Le mutazioni puntiformi sono le più comuni
- Mutazione comune: Ashkenaziti
- Delezione di 2.502-bp nell'esone 55 e parti degli introni 54 e 55
- Frequenza dei portatori: 1 su 108
- Gravità: Forma "tipica" di miopatia nemalinica
- Piccole inserzioni, delezioni o mutazioni puntiformi
- 3' fine del gene: esoni 165 ÷ 185; Parte della nebulina della linea Z
- Mutazioni non producenti la proteina nebulina
- Malattia grave
- Morte 1 giorno ÷ 9 mesi
- Insorgenza infantile: Da lieve a moderata ("Tipica")
- Insorgenza: Nascita; Presenti alcuni movimenti
- Debolezza
- Tronco
- Distale gambe: Dorsiflessori delle caviglie
- Flessori delle ginocchia
- Faccia
- Voce nasale
- Respiratoria
- Caratteristiche dismorfiche
- HEENT: Palato ad arco alto; Micrognatismo
- Contratture: Dita
- Deformità del torace
- Ipermobilità delle articolazioni
- Cardiaco: Normale
- Muscoli extraoculari: Normali
- Progressione
- Lenta o nessuna
- Può migliorare dopo una iniziale dipendenza dal respiratore
- Patologia muscolare
- Nebulina ancora presente
- Bastoncelli subsarcolemmali
- Ultrastruttura: Aberrazioni delle linee Z
- Contengono: Actinina α; Miotilina, teletonina, actina, tropomiosina e desmina
- Fibre muscolari di tipo 2: Ridotte di numero
- MRI muscolare 53
- Fenotipo lieve
- Il coinvolgimento: Tibiale anteriore, soleo
- Risparmiati: I muscoli delle coscie
- Coinvolgimento fenotipico moderato
- Retto femorale, vasto laterale, tendine del poplite, anteriore della
gamba, soleo
- Variante: Miopatia nebulinica distale
- Variante: Miopatia dei nuclei con bastoncelli
80
- Epidemiologia: 1 paziente
- Genetiche
- Recessiva
- Mutazioni della nebulina
- Duplicazioni
- Troncamento
- g.195187_195188dupAC nell'esone 140: Perdita di tutte le isoforme della nebulina
- g.234878_234881dupTCAA nell'esone 171: Perdita di alcune isoforme della nebulina
- Insorgenza
- Età: Congenita
- Ipotonia
- Insufficienza respiratoria
- Clinica
- Debolezza
- Diffusa
- Predominanza assiale
- Lieve facciale
- Non indipendente nel camminare
- Scheletrico: Scoliosi; Contrattura delle articolazioni
- Cardiaco: Normale
- Laboratorio
- EMG: Miopatica
- NCV: Normale
- Patologia muscolare: Fibre muscolari con bastoncelli e nuclei
- MRI: Anormali soleo e anteriore + compartimenti laterali; Gastrocnemio
risparmiato
- Altre miopatie dei nuclei con bastoncelli: Mutazioni RYR1
- Varianti: Altre
- Rare cause di: Forma grave congenita, o intermedia (lieve) di miopatia con bastoncelli
- Mutazioni della nebulina NON identificate nella: Miopatia con
bastoncelli ad insorgenza adulta
l
Miopatia con bastoncelli NEM3: Actina α
(ACTA1; Muscoli scheletrici)
Cromosoma 1q42.1; Dominante o
sporadica, recessiva
- Genetica
- Mutazioni
- Identificate ~15 mutazioni missenso
- Distribuite in tutti 6 esoni codificanti
- Alcune coinvolgono domini funzionali conosciuti dell'actina
- Allelica con
- Proteina actina (ACTA1)
- Actina α dei muscoli scheletrici
- Adulti normali: Presente nei muscoli ma non nel cuore
- Sviluppo: Diventa l'isoforma predominante nei muscoli al 3° trimestre di
gestazione
- Sindromi dominanti: L'ACTA1 mutante esercita un effetto negativo dominante
- L'aggregazione può essere causata da
- Shock termico
- Leptomicina B (un inibitore specifico dell'esportazione nucleare mediata
dai segnali di esportazione nucleare ricchi di leucina)
- Mutazione nei segnali di esportazione nucleare ricchi di leucina
- Clinica
- Ereditarietà dominante
- Insorgenza: Variabile all'interno delle famiglie
- Clinica: Fenotipi variabili
- Lieve
- Mutazioni: Asn115Ser; Gly268Cys; Ile136Met
- Insorgenza: Età 12
- Debolezza: Tronco e prossimale; Lieve facciale
- Insufficienza respiratoria: 5a decade
- Progressione: Lenta su decenni
- CK serica: Normale
- MRI: Coinvolgimento gluteale e coscie anterolaterali
- Morfologia muscolare
- Bastoncelli, specialmente nelle fibre sottili; Strutture simili a nuclei
- Ipertrofia delle fibre muscolari: Ile136Met
- Grave
- Nuove mutazioni dominanti: Ile357Leu; Arg183Gly
- Fenotipi
- Letale; congenito
- Debolezza congenita
- Altre mutazioni: Met132Val; Val163Met; Met269Arg
- Funzione cardiaca: Normale
- Patologia
- Anormali fibre tipo differenziazione
- glicogeno accumulazione
- Bastoncelli
- Nessuna relazione tra abbondanza e gravità della malattia
- Più nelle fibre muscolari sottili
- Colorazione per actinina α 2
- I bastoncelli tendono ad essere nelle fibre muscolari di tipo 1
- Possono essere presenti in alcuni muscoli ma non in altri
- Distruzione miofibrillare
- Verticillamento dei filamenti sottili di actina
- Ereditarietà recessiva 66
- Genetica
- Identificate mutazioni per troncamento; ? Missenso
- Troncamento: Arg41X (francesi), Tyr364fsX (spagnoli), Asp181fsX10 (britannici)
- Decorso
- Debolezza
- Grave nella maggior parte: Diffuse; Ipotonia
- Insufficienza respiratoria
- Difficoltà di alimentazione
- Morte in 1° anno: La maggior parte
- Contratture (30%)
- CK serica: Leggermente elevata o normale
- Muscoli
- Morfologia: Bastoncelli nemalinici e corpi zebrati
- Actina: Assenza di actina α scheletrica; Aumentata l'actina α cardiaca
- Lipidi: Aumentata
- Alcuni sopravvivono per tutta l'infanzia
- Pazienti con forma sporadica: Spesso mutazioni
de novo
- Variante: Miopatia nemalinica con bastoncelli intranucleari 62
- Tutte le mutazioni coinvolgono ACTA1
- Ereditarietà
- Sporadica (La maggior parte) o dominante
- Marcate variazioni fenotipiche nella famiglia
- Insorgenza: Nascita (La maggior parte) ÷ 55 anni
- Clinica
- Faccia stretta
- Muscolatura sottile
- Ipotonia
- Debolezza: Diffusa
- Laboratorio
- CK serica: Normale o leggermente elevata
- EMG: Miopatica
- Decorso
- Morte in 1° anno nel 40% dovuta a insufficienza respiratoria
- Simile alle mutazioni ACTA1 senza bastoncelli intranucleari
- Variante: Miopatia con Cap con mutazione ACTA1
82
- Epidemiologia: 1 paziente
- Mutazione: Met47Val, dominante
- Clinica
- Movimenti fetali diminuiti
- Nascita: Respirazione scarsa
- Motorio
- Ipotonia: Assiale e periferica
- Debolezza: Grave
- Movimenti spontanei minimi
- Atrofia generalizzata
- Riflessi tendinei profondi: Assenti
- Generale
- Linea dei capelli bassa
- Micrognatismo
- Palato ad arco alto
- Piega palmare singola
- Dita lunghe
- Testicoli non discesi
- Morte: 5 anni
- Laboratorio
- CK serica: Normale
- MRI del capo: Normale
- Biopsie muscolari
- 5 settimane: Tipo 1 assottigliate; No Cap
- 4 anni
- Cap: Contengono filamenti sottili, actinina α,
actina e desmina; NADH+
- Nuclei interni
- Linee Z espanse
- MRI muscolare
- Segnali muscolari coinvolti: T1 aumentato
- Muscoli coinvolti: Diffuso coinvolgimento della coscia e della gamba
- Relativamente risparmiato: Gastrocnemio
l
Miopatia con bastoncelli NEM4
: Tropomiosina β (TPM2)
Cromosoma 9p13; Dominante
- Mutazioni
- Missenso Q147P e E117K
- Le mutazioni in TPM2 causano anche
- Proteina tropomiosina β
- Frequenza: Rara
- Insorgenza: Infanzia ÷ fanciullezza
- Clinica
- Debolezza: Prossimale > distale; Alcuni con Insufficienza respiratoria
- Contratture: Qualche o nessuna
- Progressione: Lenta o nessuna
- Variante: Sindrome di Escobar
74
- Epidemiologia: Famiglia algerina consanguinea
- Genetica
- Ereditarietà: Recessiva
- Mutazioni
- Omozigotiche
- Gln210Stop: Esone 6b, TM β isoforma 1 specifica dei muscoli scheletrici
- Proteina TPM2: Assenza della isoforma dei muscoli scheletrici della tropomiosina β
- Clinica
- Insorgenza: Congenita
- Motorio: Ipotonia; Pietre miliari ritardate
- Sofferenza respiratoria
- Articolazioni: Pterigia; Artrogriposi
- Cardiaco: Blocco ventricolare bifascicolare con blocco del fascio destro
ed emiblocco del sinistro
anteriore
- Muscoli
- Tessuto connettivo: Abbondante
- Bastoncelli: Più comuni in fibre muscolari di tipo I (60%)
l
Miopatia con bastoncelli NEM5
:
Troponina T1 (Scheletrico, Lento;
TNNT1)
13
Cromosoma 19q13.4; Recessiva
- Mutazione: Nonsenso; E180X; Omozigotica
- Epidemiologia: Amish del vecchio ordine
- Clinica
- Insorgenza
- 1° mese di vita
- Tremori
- Inizio entro pochi giorni dalla nascita
- Distribuzione: Mascella e arti inferiori
- Si risolvono entro 2 ÷ 3 mesi
- Ipotonia
- Contratture: Spalle e anche; Lievi
- Progressione
- Contratture prossimali: Aumentano
- Debolezza: Prossimale > distale
- Petto carenato: Gabbia toracica rigida; Sviluppo di contratture prossimali
- Cardiaco: Non 1° coinvolgimento; Insufficienza congestiva terminale destra
- Morte per insufficienza respiratoria < 2 anni
- Patologia
- Sproporzione delle fibre di tipo I
- Inizianti dalla linea Z
- Bastoncelli: Situati centralmente
- Distruzione miofibrillare
- Degenerazione delle miofibre
l
Miopatia con bastoncelli con movimenti lenti NEM6
27
Cromosoma 15q21-q23; Dominante
- Genetica: Nessuna mutazione TPM1
- Insorgenza: Fanciullezza
- Clinica
- Debolezza: Lieve; Prossimale e flessori del collo
- Movimenti: Lenti; Incapacità a correre; Scarsa correzione posturale rapida
- Progressione: Lenta
- Scheletrico (Alcuni pazienti): Palato alto; Deformità toraciche
- Laboratorio
- EMG: Miopatica
- CT muscolare: Infiltrazione di grassi nei muscoli prossimali degli arti
e troncali
- Biopsia muscolare
- Predominanza di tipo I
- Nuclei interni
- Bastoncelli: Granulari; Diffusamente distribuiti; Tutte le fibre
- Strutture simili a nuclei
- Contorni meno definiti che nei nuclei tipici
- Ultrastruttura: Grandi regioni di bastoncelli con perdita della struttura muscolare
l
Miopatia con bastoncelli NEM7
: Cofilina 2
(CFL2)
 65
65
Cromosoma 14q12; Recessiva
- Epidemiologia: 1 famiglia medio orientale, 1 fratello e 1 sorella
- Genetica
- Mutazione: Missenso, Ala35Thr; Omozigotica
- Eterozigoti: Non affetti
- Proteina cofilina-2
- Membro
del gruppo delle proteine AC (ADF/Cofilina)
- Regola le dinamiche dei filamenti di actina: Fattori di
depolimerizzazione dell'actina
- Altri membri del gruppo: Cofilina 1
; Destrina
- Isoforma specifica dei muscoli scheletrici
- Localizzata nei filamenti sottili
- Esercita effetto sull'actina, in parte, attraverso interazioni con
le tropomiosine
- Lega le actine G e F in rapporto 1:1
- Maggior componente dei bastoncelli di actina intranucleari e
citoplasmatici
- Clinica
- Insorgenza: Ipotonia alla nascita
- Prime pietre miliari motorie: Ritardate
- Deambulazione: Presente per brevi distanze; Cadute frequenti; Incapacità a correre
- Non debolezza facciale o piedi ricadenti
- Patologia muscolare
- Bastoncelli nemalinici: 1 dei 2 pazienti
- Mininuclei: Qualche fibra
- Predominanza delle fibre muscolari di tipo I
- Variabilità nelle dimensioni delle fibre
- Corpi laminati concentrici
- Cofilina 2
- Colorazione sarcomerica: Ridotta
- Forme fosforilate: Assenti
- Colorazione falloidina: Il 4% delle miofibre contengono accumulazioni di
filamenti di actina
l
Miopatia con bastoncelli: Lieve
64
Dominante
- Epidemiologia: Famiglia svizzera
- Insorgenza
- Clinica: Decorso lieve
- Camminare: Inizio 18 ÷ 24 mesi
- Debolezza: Assiale; e prossimale
- Difficoltà a correre
- I più senza limitazioni nei normali ADL
- Biopsia muscolare
- Predominanza delle fibre muscolari di tipo 1
- Bastoncelli: 1% ÷ 5% delle fibre muscolari; Aumenta con l'età
l
Altre forme di miopatia a bastoncelli
- Tipi ad insorgenza infantile: Gravi
- Sequenza della acinesia fetale
- Insorgenza: 2° trimestre di gravidanza
- Polidramnio; Contratture delle articolazioni (Multiple); Ipoplasia
polmonare
- Non collegata a actina α, nebulina o tropomiosina α 3
- Nascita
- Patologia: Bastoncelli citoplasmatici e intranucleari
- Può essere necessaria la lente ad immersione in olio visualizzarli
- Miopatia con bastoncelli ad insorgenza adulta: Eterogenea
59
- Spesso senza storia familiare
- Insorgenza
- Clinica
- Debolezza
- Mialgia
- Cardiomiopatia
- Progressione
- Alta mortalità nel 40%: Morte entro 1 anno
- Lenta progressione per anni in altri casi
- Laboratorio
- Biopsia muscolare
- Bastoncelli
- Immunocolorazione per actinina α o miotilina
- I bastoncelli possono essere visti meglio con la tricromia in
sezioni di 3 μM
- Le fibre contenenti bastoncelli hanno anche
- Grandi nuclei vescicolari
- Basofilia citoplasmatica focale
- Vacuoli piccoli
- Infiammazione (30%)
- Fibre muscolari necrotiche: Occasionali
- Fibre muscolari lobulate (50%)
- Predominanza di tipo I (20%)
- Ultrastruttura: Bastoncelli < 1μM in lunghezza
- Proteina M (50%): ? Prognosi sfavorevole
- CK serica: Normale o leggermente elevata
- EMG: Miopatia irritabile
- Escludere
- Trattamento: Incerto; ? Prednisone
- Malattia del nucleo centrale, con bastoncelli e ipertermia maligna
Miopatia Centronucleare (Miotubulare)
- Miopatia centronucleare: Collegata a X (MTM 1)
l
Miotubularina (MTM1)
; Cromosoma Xq27.3-q28;
Recessiva
- Epidemiologia: 1 maschio ogni 50.000
- Caratteristiche genetiche
- 15 esoni: La traslazione inizia dal codone nell'esone 2
- Espressione genetica
- Ubiquitaria
- Trascrizione più piccola espressa selettivamente nei muscoli scheletrici e
nei testicoli
- Ha caratteristiche di funzione domestica
- Alto contenuto GC del promotore
MTM1
- Non definite le strutture box TATA o box CAAT
-
Mutazioni genetiche: Diffuse in modo non uniforme attraverso l'intero gene
- Generale
- In 248 famiglie identificate 151 malattie differenti correlate a mutazioni
10
- Le mutazioni sono ampiamente distribuite nell'intero gene
- Punti caldi: 6 mutazioni ricorrenti
- Frameshift nel codone 38; P205L; R224X; R241C; 420insFIQ; R421X
- Arg241 Cys: 2a mutazione più frequente; Causa miopatia lieve (72%) o
grave
- Mutazioni nei maschi > femmine
- Tipi di mutazione
- Mutazioni puntiformi: Missenso (55) e Nonsenso (40); La maggior parte
delle tra gli esoni 8 e 12
- Piccole inserzioni o delezioni (50)
- Grandi delezioni (15)
- Mutazioni nel sito di splice (38)
- IVS11, A-G, -10: La mutazione più comune; Causa grave
miopatia
- In alcune famiglie identificato mosaicismo della linea germinale
- Localizzazione delle mutazioni
- Maggior frequenza negli esoni: 4 > 12 > 3 > 8 > 9 > 11
- Regioni altamente conservate
- ? Riflette l'importanza delle regioni mutate del gene sui domini
funzionali
- Correlazioni genotipo-fenotipo
- Mutazioni per troncamento e sito di splicing
- Solitamente associate con fenotipo grave
- Frequenti morte o dipendenza dal ventilatore
- Dimensione delle fibre muscolari: Più sottili
- Alcuni fenotipi lievi con mutazioni molto distali
- Mutazioni missenso e di delezione di singolo aminoacido
- Più alta prevalenza di fenotipo lieve (33%)
- Tutti in
siti di fosfatasi attiva o dominio SID hanno un fenotipo grave
- Alterazioni della proteina
- Prevista grave: 99% con fenotipo grave
- Livello normale della miotubularina: Mutazione missenso o piccola delezione o
inserzione
- Delezione cromosomica grande: Associata ad anormale sviluppo genitale
- Alcune femmine sintomatiche portatrici
- Molte mutazioni possono essere spiegate con
- Riparazione del DNA mediata da metilazione dei dinucleotidi CpG
modificati, o
- Slippaggio della DNA polimerasi con brevi sequenze ripetute in tandem
- Caratteristiche della proteina
MTM1
- Localizzazione tissutale: Espressa ubiquitariamente
- Localizzazione cellulare
- Sarcolemma
- Banda I, triadi comprese
- Associata con gli endosomi
- ? Nucleare
- Struttura della MTM1
- Contiene il sito attivo della proteina tirosina fosfatasi (PTP)
- Mostra similarità alla doppia specificità della serina/tirosina fosfatasi
(dsPTPases)
- Funzioni della MTM1
- Azione enzimatica: Defosforila fosfatidilinositolo 3-fosfato
- Associata con i domini SET (Suvar3-9, potenziatore di zeste, Trithorax)
- Domini SET: Nelle proteine contribuiscono ai meccanismi epigenetici
della regolazione genetica
del gene
- ? Ruoli: Maturazione delle fibre muscolari;
Omeostasi della membrana plasmatica
- Sovraesspressione
- Induce la formazione di proiezioni simili a filopodi
- Colpisce il successivo passaggio dagli endosomi ai lisosomi
- Muscoli
- Proliferazione delle strutture della membrana che contengono miotubularina
- Vacuoli contrassegnati dai marcatori del sarcolemma e dei tubuli T (caveolina 3,
distrofina, DHPR)
- Famiglia della miotubularina famiglia e disturbi correlati
- Proteina correlata alla miotubularina 2: CMT 4B
- SBF2 (MTMR13): CMT 4B2
- Fosfoinositidi endosomici: PIP5K3
; Distrofia della cornea
a macchie di François-Neetens
- Caratteristiche cliniche
- Insorgenza: Infanzia
- Polidramnios: 50%
- Scheletrico: Capo grande (70%); Faccia stretta (80%); Dita lunghe (60%)
- Motorio
- Ipotonia grave
- Debolezza: Prossimale e distale; Simmetrica
- Insufficienza respiratoria
- Oftalmoplegia e ptosi
- Prognosi
- Morte precoce: Media = 5 mesi
- ? Miglioramento dopo periodo neonatale
- Debolezza non progressiva
- Coloro che sopravvivono dono spesso dipendenti dal respiratore (80%)
- Caratteristiche sistemiche in alcuni sopravissuti > 1 anno
- Stenosi pilorica
- Sferocitosi
- Calcoli biliari
- Renale: Calcoli o calcinosi
- Diatesi sanguinolenta: Rispondente alla vitamina K
- Scheletrico: Rapida crescita lineare; Età ossea avanzata
- Disfunzione epatica
- Laboratorio
- CK: Normale o lievemente elevata
- EMG
- Potenziali delle unità motorie: Piccola ampiezza; Polifasici
- Attività spontanee: Fibrillazioni; Picchi d'onda positivi; Scariche ripetitive complesse
- Patologia muscolare 67
- Nucleo centrale singolo
- In alcune fibre muscolari: Gamma = 4% ÷ 40%
- La frequenza può aumentare con l'età
- Predominanza delle fibre muscolari di tipo 1
- Dimensione delle fibre muscolari
- Dimensione media delle fibre: Ridotta
- Aumento in dimensioni con l'età: Ridotto
- Le fibre muscolari di tipo 1 più sottili di quelle di tipo 2: Più prominente
nei pazienti più anziani
- Dimensione delle fibre più piccola nella malattia più grave
- Dimensione delle fibre più piccola con mutazioni di troncamento/delezione
che con le mutazioni missenso
- Miotubularina mutazione: Portatori
- Occasionali: Debolezza facciale lieve
- Più grave
56
- Genetica
- Mutazioni: R253X; c.1354–1G>A (Sito di splicing)
- Inattivazione di X distorta: 1 famiglia
- ? Altri meccanismi
- Penetranza incompleta
- Insorgenza
- 1a decade
- Debolezza delle braccia
- Disturbi dell'andatura
- Asimmetria scheletrica
- Debolezza
- Iniziale: Braccia
- Prossimale e distale: Con la progressione della malattia
- Emidiaframma elevato
- Progressiva dalla 3a alla 5a decade
- Scheletro
- Asimmetria
- Cifoscoliosi
- Piede equino (Bilaterale)
- CK serica: Normale
- Patologia muscolare
- Dimensione delle fibre variata: Entrambi tipi di fibre
- Nuclei interni
- NADH: Normale architettura interna
- Neonatale onset39
- Mutazione: 605delT
- Casi non familiari
- Clinica
- Ipotonia
- Lassità delle articolazioni
- Andatura anserina
- Debolezza: Dei cingoli degli arti; Facciale
- Riflessi tendinei: Assenti
- Miotubularina topo knock-out 37
- Debolezza progressiva
- Sviluppo delle fibre muscolari
- Inizialmente normale con nuclei periferici
- Successivamente: Alterazioni degenerative e nuclei centrali
- Patologia delle fibre muscolari
- Variabile fra i muscoli: Più nei bicipiti
- Nelle fibre muscolari di tipo I e II
- Atrofia
- Localizzazione sbagliata di mitocondri e nuclei
- CK serica: Leggermente elevata
- Tessuti diversi dai muscoli: risparmiati
- Miopatia centronucleare: Autosomica recessiva
- Generale
- Insorgenza
- Infanzia, fanciullezza, adulti < 30 anni: ~1/3 ognuna
-
Possibili sottogruppi
49
- Insorgenza precoce con oftalmoplegia
- Insorgenza precoce senza oftalmoplegia
- a tarda insorgenza senza oftalmoplegia
- Clinica
- Debolezza: Prossimale > distale
- Oftalmoplegia: Pazienti con insorgenza precoce
- Debolezza facciale
- Solitamente sopravvivenza dopo l'infanzia
l
Anfifisina 2 ( Integratore ponte 1 (Bridging)(BIN1))
; Cromosoma 2q14;
Recessiva
68
- Epidemiologia: 4 individui di 3 Famiglie consanguinee; India e Iraq
- Genetica: Mutazioni
- Tipi: Missenso e stop; K35N, D151N, K575X
- Effetto: Tutte provocano solamente una parziale perdita di funzione
- Omozigotiche
- Proteina BIN1
- Coinvolta nel rimodellamento della membrana
- Regolato dagli fosfoinositidi
- Ruolo nell'organizzazione dei tubuli T
- Mutazioni
- Perdita del collegamento funzionale alla dinamina 2
- ? Biogenesi disordinata dei tubuli T
- Cellule maligne: Spesso ridotte
- Clinica
- Insorgenza
- Età: Dalla nascita alla fanciullezza (8 anni (mutazione D151N))
- Gravidanza: Movimenti fetali ridotti o normali
- Debolezza
- Prossimale
- Lieve
- Oftalmoparesi e ptosi: Alcuni pazienti
- Nessuna insufficienza respiratoria
- Contratture: Nascita
- Cardiaco: Dilatazione in alcuni pazienti
- Decorso
- Debolezza lentamente progressiva
- Sopravvivenza oltre l'infanzia: 60%
- Patologia
- Dimensioni delle fibre: Minore variabilità che in altri tipi di CNM
- Tipi di fibre
- Popolazione omogenea di fibre di tipo I ipotrofiche arrotondate
- Predominanza di tipo I
- Nuclei centrali
- In numerose fibre
- Numerosi nuclei nella parte centrale delle fibre
- NADH
- Nuclei centrali (regione chiara) circondati da un bordo di materiale
densamente colorato
- Organizzazione radiale del reticolo sarcoplasmatico: Non presente
- Tessuto connettivo endomisiale: Aumentato
- Necrosi e rigenerazione: Non presente
- Caveolina 3: Immunoreattività in alcune fibre in vacuoli localizzati
centralmente
- Recettore a della diidropiridina marcatore dei tubuli T (DHPRa):
Immunoreattività attorno a nuclei localizzati centralmente
Miopatia centronucleare: Autosomica dominante
l
MYF6
; Cromosoma 12q21;
Dominante
- Epidemiologia: 1 paziente
- Mutazione genetica MYF6: Ala112Ser
- Proteina MYF6
- Fattore di determinazione muscolare
- Dominio elica--ansa--elica: Comune caratteristica dei fattori miogenici
- Clinica
- Insorgenza: Tarda fanciullezza ÷ adulti
- Debolezza: Prossimale > distale
- Crampi alle gambe
- ± Oftalmoplegia, debolezza facciale o scapolare
- Mutazione MYF6 insieme alla distrofia di Becker: Converte la malattia
nel fenotipo più grave
- Laboratorio
- CK: Normale o leggermente elevata
- Biopsia muscolare: Miopatica; Fibre inanellate; Nuclei centrali
l
Dinamina 2 (DNM2)
; Cromosoma 19p13.2;
Dominante o sporadica 60
- Epidemiologia: Molte famiglie con miopatia centronucleare
- Genetica
- Mutazioni puntiformi: R465W; R369Q; R369W; E368K
- Localizzazioni
- Esoni 8 e 11
- Dominio medio: Coinvolto nell'auto assemblaggio e nella
localizzazione centrosomica della proteina
- Allelica con
- Proteina DNM2
- Espressa ubiquitariamente
- Associata con i microtubuli
- Famiglia: Grandi GTPasi
- Lega a BIN1 e SNX9
- Implicata nella endocitosi e nella motilità cellulare
- Regola l'invaginazione tubulare della membrana plasmatica
- Parte dell'apparato di fusione e fissione cellulare
- Insorgenza
- Età
- Prime pietre miliari: Normali
- Solite età: Adolescenza e adulti
- ? Mutazione R465W con insorgenza più precoce
- Sintomi
- Dolore muscolare correlato all'esercizio
- Debolezza: Difficoltà a camminare ed a salire le scale; Cadute; Scarse prestazioni sportive
- Clinica
- Debolezza
- Distale > prossimale: Estremità superiore tutte le distali
- Assiale: Flessori del collo; Addome
- Oculare: Ptosi; Sguardo verso l'alto ridotto (20%)
- Faccia 40%
- Respiratorio: Normale
- Progressione: Lenta; 1 paziente in carrozzina nella 6a decade
- Contratture: Caviglie (90%); Dita
- Altre alterazioni descritte in qualche paziente
- Retinopatia
- Cardiomiopatia: In femmine portatrici
- Laboratorio
- CK serica: Normale o leggermente alta
- NCV: CMAP ridotti; Alcune NCV lievemente lente
- EMG: Miopatica; Alcuni con attività spontanea
- MRI: Precoce coinvolgimento delle gambe distale posteriore
- Coinvolti: Soleo laterale, gastrocnemio; Coscia posteriore; Adduttore lungo; Bicipiti; Semimembranoso
- Risparmiati: Sartorio, gracile e retto femorale
- Muscoli
- Nuclei centrali: Alcuni a catena
- NADH: Alterazioni "simili a raggi"
- Regione centrale delle fibre in regioni senza nuclei Colorazione per: PAS,
fosforilasi ed enzimi ossidativi
- Lesioni simili a nuclei nei muscoli prossimali più preservati
- Nessun multi-mininucleo
- Ultrastruttura
- I nuclei centrali sono normali
- Gli spazi centrali senza nuclei hanno: Mitocondri, Reticolo sarcoplasmatico,
Complesso di Golgi e particelle di glicogeno
- Distribuzione radiale dei filamenti sarcoplasmatici intermiofibrillari
- Variante: CNM neonatale grave con prognosi favorevole
71
- Storia familiare: Sporadica o dominante
- Genetica: Mutazioni
- Eterozigotiche
- Esone 16: Dominio con omologia alla plecstrina, regioni dei terminali N e C
- Tipi: Missenso o in frame
- Ala618Thr; A618D; Ser619Leu; Ser619Trp; V625del
- Insorgenza
- Clinica
- Gravidanza: Normale
- Ipotonia; Suzione debole
- Nervi cranici: Faccia; Ptosi; Oftalmoparesi
- Debolezza: Generalizzata; Distale > prossimale
- Respiratorio: Sindrome restrittiva in
alcuni pazienti
- Funzione cardiaca: Normale
- Nervi periferici: Normali
- Prognosi: Favorevole; Lento miglioramento dalla media infanzia o da adulti
- Laboratorio
- CK serica: Normale a leggermente aumentata
- EMG: Miopatica
- muscolo
- Predominanza di tipo I e assottigliate
- Nuclei centrali (3% ÷ 50%): In entrambi i tipi di fibre
- Fibre grosse: Può essere di entrambi i tipi
- Filamenti sarcoplasmatici radiali: Alcuni
- Variante: CNM con cataratte
- Mutazione: Leu621Pro
- Clinica
- Insorgenza: Neonatale
- Ipotonia
- Insufficienza respiratoria
- Cataratte
- Oftalmoplegia esterna
- Decorso: Progressivo; Morte a 14 anni
l
Miopatia centronucleare: Fenotipi geneticamente indefiniti
- Sottotipo clinico 1:
Miopatia
centronucleare tipica dominante
49
- Epidemiologia: 2 famiglie
- Età di insorgenza: Fanciullezza a adulta
- Clinica
- Debolezza
- Gravità: Lieve
- Distribuzione: Diffusa; Prossimale e distale; Occasionali predominanza distale
- Progressione: Lenta
- Disabilità: Lieve; La maggior parte deambulanti fino alla 6a
decade
- Ptosi: La maggior parte
- Contratture: Qualche paziente
- Tremore: Posturale; Alcuni pazienti
- Laboratorio
- CK serica: Normale
- ECG: Normale
- Funzione polmonare: Normale o ridotta
- EMG: Miogenica
- Sottotipo clinico 2: Miopatia centronucleare tipica dominante con
ipertrofia muscolare
49
- Epidemiologia: 1 famiglia
- Insorgenza: Adolescenza e adulti, Rara infantile
- Clinica
- Ipertrofia muscolare: Diffusa
- Debolezza
- lentamente progressive
- Braccia distali
- Oculare
- Ptosi: Comune
- Oftalmoplegia: Occasionali pazienti
- Scheletrico
- Spina rigida: Lieve 40%
- Contratture alle caviglie: 30%
- Contratture delle dita: 14%
- Laboratorio
- CK serica: Alta (2x ÷ 4x) in alcuni pazienti
- EMG: Miopatica
- FVC: Può essere ridotta
Miopatia centronucleare: Biopsia muscolare
- Nuclei centrali
- Pallore centrale alla ATPasi
- Fibre di tipo 1 predominanti e sottili (50%)
- NADH: Distribuzione radiale dei filamenti sarcoplasmatici
- Il 50% dei trasportatori ha nuclei centrali nelle fibre muscolari
- Fibrosi endomisiale: Più grave nei casi recessivi
Miopatia centronucleare: Cane 50
l
Cromosoma canino 2
Distrofie muscolari congenite
Distrofia muscolare congenita: Caratteristiche tipiche
- Ereditarietà: Autosomica recessiva (AR)
- Frequenza: Comune causa di disturbi neuromuscolari AR
- Insorgenza: Congenita o Infanzia (< 1 anno)
- Caratteristiche cliniche
- Debolezza: Diffusa
- Contratture
- CNS
- Comune nelle forme gravi di CMD
- Può essere subclinica
- Disturbi della mielina o nella migrazione neuronale
- Patologia dei muscoli
- Miopatica
- Dimensione delle fibre variata
- Aumento del tessuto connettivo endomisiale
- Degenerazione delle fibre muscolari sottili dopo 4 anni
- Disturbi delle molecole del tessuto connettivo
- Laminina α2:
MDC1A
- Collagene, tipo VI: Ullrich
- Disturbi
della glicosilazione
48
- Molecole marcatrici: Distroglicano α
- Struttura comune del mannosile glicano O:
NeuAcα2-3Galα1-4GlcNAcα1-2Man-Ser/Thr
- Disturbi: Mannosilazione O difettosa del distroglicano α
- Disturbi specifici
Distrofia muscolare congenita di Fukuyama
l
Fukutina (FKTN)
; Cromosoma 9q31;
Recessiva
- Epidemiologia
- Giappone: Comune; L'incidenza è del 40% delle MD di Duchenne
- Paesi occidentali: Rara
- Genetica
- Mutazioni
- 80% dei pazienti giapponesi hanno un aplotipo simile
- Inserzione
retrotrasposizionale
(3kb) di sequenze ripetute in tandem (genotipo ancestrale)
- Localizzazione: 3' fine non traslata del gene fukutina
- Effetto della mutazione: Trascrizione della fukutina ridotta
- Fino al 100% dei pazienti giapponesi sono almeno eterozigotici per
l'inserzione ripetuta in tandem (genotipo ancestrale)
- Altre mutazioni
- Pazienti giapponesi: Codoni di stop; Mutazioni puntiformi e piccole
delezioni
- Paziente turco: Omozigotico per inserzione di 1 bp (nt504(insT)) nell'esone 5
- Ashkenaziti 76
- Mutazione: c.1167insA; Omozigotica
- Portatori: 0,7% degli ashkenaziti americani
- Fenotipo: Grave
- Correlazioni genetiche e cliniche
- Fenotipo più lieve: Omozigotico per retrotrasposone
- Fenotipo grave: Eterozigotico per retrotrasposone e mutazione puntiforme
- Maggiore coinvolgimento oculare
- Omozigosità per mutazioni puntiformi
- Riportata sindrome simile a WWS
- Può essere letale
- Allelica con
-
Proteina fukutina 17
- mRNA nel cervello
- Espressa in livelli similari in adulti e nel Cervello fetale
- Adulti: Neuroni corticali
- Fetale: Precursori neuronali e cellule migranti
- Localizzazione cellulare: Neuroni; Non negli astrociti
- Proteina
- ? Più abbondante nel Cervello fetale
- Probabilmente secreta
- Muscoli scheletrici: Non rintracciabile con l'immunocitochimica
- Clinica
- Generale
- Cervello: Coinvolgimento più lieve che nelle Walker-Warburg
o muscoli-occhi-cervello
- Occhi: Solo occasionalmente gravemente colpito
- Eterogeneità clinica: Alcuni pazienti camminare e hanno una sopravvivenza più lunga
- Caratteristiche cliniche: Casi gravi
- Deficit motori
- Insorgenza: Mesi ÷ 1 anno
- Ipotonia: Grave
- Debolezza e atrofia: Faccia e Arti
- Spasticità: Occasionali
- Faccia
- Scheletro
- Contratture
- Cifoscoliosi
- Cranio: Microcefalia; Asimmetria
- CNS
- Attacchi epilettici (50%)
- Ritardo mentale: Grave; IQ 30 ÷ 50
- Idrocefalo: Progressivo
- Oculare
- Miopia
- Ipoplasia e distacco retinici
- Strabismo
- Persistenza dei corpi vitrei primari e dell'arteria ialoide
- Microftalmo
- Atrofia del nervo ottico
- Cecità: Malformazioni delle camere oculari anteriore e posteriore
- Decorso
- Disabilità progressiva
- Morte: Spesso < 11 anni; Disfunzione respiratoria
- Caratteristiche cliniche: Casi più lievi o tipici
- Tipico picco della funzione motoria: Seduta senza assistenza
- I casi più leggeri: Camminata senza assistenza
- Laboratorio
- Patologia del CNS
- Corteccia
- Difetti migrazionali
- Migrazione neuronale radiale e tangenziale alterata
- Micropoligiria (lissencefalia di tipo II), acciottolato
- Assenza di laminazione nella materia grigia
- Interruzioni nella glia limitante e nella lamina basale
- mRNA fukutina nel cervello con CMD di Fukuyama
- Assente nelle regioni di displasia sovramigrata
- Moderate espressione nelle aree più normali
- Materia bianca
- Ipomielinizzazione
- Assenza del corpo calloso e del setto pellucido: Alcuni pazienti
- Scansione CT: Luminosità, specialmente frontali, Regione occipitale risparmiata
- Fossa posteriore
- Tronco cerebrale: Distorsioni
- Ponte: Appiattimento
- Cerebello: Ipoplasia
- Encefalocele posteriore
- Pazienti più anziani
- Percorsi cerebellari efferenti: Nucleo dentato, peduncolo cerebellare superiore e nucleo rosso
- Nucleo talamico laterale
- CK serica: Moderatamente alta
- EMG: Miopatica
- Patologia muscolare:
Miopatica
- Tessuto connettivo: Aumentato
- Dimensioni delle fibre muscolari: Variabili
- Nuclei interni
- Coinvolgimento differenziale dei fasci muscolari
- Lamina basale: Distrutta
- Proteine muscolari
- Distroglicano α: Grandemente ridotto nella membrana superficiale e
nelle giunzioni neuromuscolari
- Laminina α2: Ridotta
- Sindrome variante
Distrofia muscolare congenita: Carenza di merosina
(catena della laminina α2)
l
Laminina α2; Cromosoma 6q22; Recessiva
- Mutazioni nella catena della laminina α2
- Delezione, codone di stop e occasionale missenso
- La mutazione più comune (23%): Delezione di 2 paia di basi @
2.096÷2.097
- Malattia lieve: Mutazioni
- Delezione interna alla frame: Proteina piccola (mancante del terminale N)
- Mutazioni nella sequenza di consenso conservata del donatore di splicing
degli introni 37 e
63
- Mutazioni insolite: Nel dominio o in quello simile a bastoncino
- Espressione della merosina
- Grave: Subunità anormali da 300 kDa e 80 kDa
- Lieve o ad insorgenza più tardiva: Subunità da 300 kDa spesso ridotta
selettivamente
- Prole dei portatori: Eterozigoti più comuni della probabilità prevista
- Difetto nello stesso gene nel topo dy
- Proteina laminina α2 (Merosina)
- Glicoproteina eterotrimerica
- Catena pesante (400 kDa): Catena della proteina α2
- Catene leggere (200 kDa): β1 e γ1
- Localizzazione
- Membrana basale
- Muscoli, cute, CNS e nervi periferici (Cellule di Schwann)
- Lega al distroglicano α nella lamina basale
- Laminina α2: Proteina da 400 kD
- Suddivisa dopo la traslazione in subunità da 300 kD e 80 kD
- La subunità rimangono associate dai legami disolfuro
- Il dominio globulare attaccato al distroglicano α
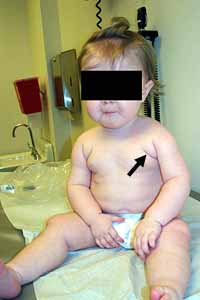
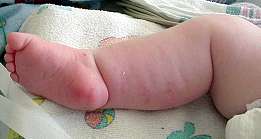
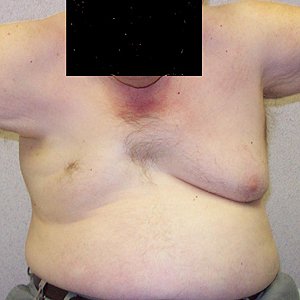
Pieghe pettorali (Freccia) con
grave debolezza delle spalle
|
- Distrofia muscolare congenita: Caratteristiche cliniche
- Debolezza
- Correlata con il livello della proteina laminina α2 residua
- Assenza della proteina laminina α2
- Debolezza grave
- Distribuzione: Simmetrica; Prossimale e Distale; Faccia
- Non progressiva
- Laminina α2 ridotta: Vedi sindromi varianti
- Muscoli: Atrofia e ipotonia
- Deambulazione
- Malattia grave: Mai
- Malattia lieve: A 2-3 anni
- Contratture
- Malattia lieve: Rigidità delle caviglie
- Malattia grave
- Contratture a molteplici articolazioni
- Anche; Ginocchia; Caviglie; Gomiti; Estensori del collo
- Progressive con l'aumentare dell'età
- CNS
- Intelligenza spesso normale
- In alcuni disfunzione motorio percettiva
- Attacchi epilettici (20%)
- Neuropatia: Lieve; Occasionale; ±
demielinizzazione
- Decorso
- Contratture progressivamente gravi
- Malattia grave: Morte 15 ÷ 30 anni 2°
ad insufficienza respiratoria
- Laboratorio
- CK: Moderatamente alta
- MRI del CNS: Alterazioni della materia bianca (Segnale su T2 aumentato); Corteccia
solitamente normale
- Disturbi della laminina α2: Patologia muscolare
- Colorazione merosina (Laminina α2)
- Solitamente assente: 95% con assenza di merosina hanno mutazione genetica
- Perdita
parziale
di merosina: Disabilità più lieve o insorgenza più tardiva
- Clinica: Disabilità più lieve o insorgenza più tardiva; Attacchi
epilettici ad insorgenza tardiva
- Patologia muscolare: Miopatia; In alcuni vacuoli bordati e infiammazione
- Ridotta specialmente la subunità da 300 kD
- Correlata con la riduzione della catena della laminina β2
- Occasionali 2° a perdita in altre sindromi
- Normali i potenziali visivi evocati
- Patologia: Dipendente dall'età
- Fanciullezza: Fibre di dimensioni variabili; Aumento del tessuto connettivo endomisiale
- Neonatale : Può essere "predistrofica" con solamente alcune fibre
muscolari degeneranti ed in in rigenerazione
- Infiammazione: Rara, eccetto nelle biopsie da pazienti molto giovani
- Malattia lieve (perdita parziale di merosina)
- Distrofica: Variazione nella dimensione delle fibre Tessuto endomisialconnettivo aumentato
- Altre: Nuclei interni aumentati; Occasionali rotture, fibre
attorcigliate e ipercontratte
- Disturbi della laminina α2: Sindromi varianti
- Laminina α2
ridotta (perdita del terminale N da 300 kD)
- Genetica: Probabilmente allelica con i disturbi della laminina α2
- Insorgenza: Fino a 12 anni
- Debolezza
- Debolezza lieve
- Prossimale ± Distale; Simmetrica
- Si può avere ipertrofia del polpaccio
- Decorso: ? Progressivo
- Si può avere leucoencefalopatia
- Patologia muscolare
- Miopatia
- Vacuoli bordati
- Infiammazione in alcuni
- Laminina α2 ridotta: Perdita del terminale N da 300 kD
-
Neuropatia ipermielinizzante; Ritardo mentale; Epilessia
31
- Genetica: Probabilmente allelica con i disturbi della laminina α2
- Insorgenza: Fanciullezza; Ritardo nello sviluppo
- Clinica
- Debolezza: Distale; Piedi > mani
- Ritardo mentale: Moderato
- Epilessia refrattaria: Assenza ± miocloni palpebrali; Attacchi epilettici parziali, tonici e complessi
- Riflessi tendinei: Assenti
- Nessuna atrofia muscolare o contratture delle articolazioni
- Laboratorio
- CK serica: Alta; 530
- Velocità di conduzione nervosa: Ridotta
- MRI del cervello: Diffuso coinvolgimento della materia bianca
- Biopsia muscolare
- Miopatia
- Caratteristiche neurogene
- Laminina α2 lievemente ridotta
- Biopsia dei nervi
- Laminina α2 virtualmente assente
- Ipermielinizzazione ‘Globulare’: Localizzata a regioni paranodali
- Lieve perdita di assoni mielinizzati
- Nessuna alterazione di demielinizzazione-remielinizzazione cambiamenti
- LGMD 2I
- Genetica: Mutazioni FKRP con riduzione della laminina α2 nei muscoli
Modelli di topo
- dy/dy
- dy2J/dy2J: Fenotipo più lieve; Subunità da 300kD
ridotta selettivamente
Merosina normale : Forma "pura" della MD congenita
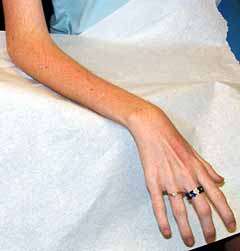
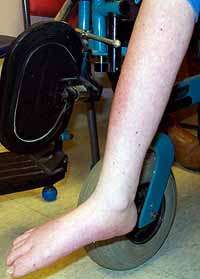
CMD: Normale merosina
|
l
Recessiva
- Geni e sindromi
- Caratteristiche cliniche
- Debolezza da lieve a moderata
- Decorso non progressivo; Disabilità lieve
- Contratture
- Intelligenza normale
- Spina rigida
- CK: Da normale a moderatamente alta
- MRI: CNS normale
- Muscoli
- Alcuni pazienti hanno assenza di α actinina-3
nei muscoli
- Non identificati difetti genetici nell'actinina
MD congenita con
mutazioni dell'integrina α 7
l
Integrina α-7 (ITGA7)
; Cromosoma 12q13;
Recessiva
- Epidemiologia: 3 pazienti giapponesi
- Alterazione del gene: Delezione di 98 paia di basi la più comune
- Proteina integrina α7β1
- Subunità con specificità tissutale
- α7: Specifica muscolare
- β1: Espressa in molti tessuti
- Abbondanza: L'integrina α7β1è l'integrina principale nei muscoli
- Funzioni
- Recettore della laminina
- Collegamento transmembrana: Dal citoscheletro
alla
matrice extracellulare
- Migrazione dei mioblasti
- Formazione di
- Giunzioni miotendinee
- Membrane postsinaptiche
- Alterazioni in altre miopatie
- Clinica
- Motorio
- Ipotonia
- Pietre miliari ritardate
- Camminata: Alcuni pazienti; 2 ÷ 3 anni
- Debolezza prossimale
- Coinvolgimento respiratorio: Con la progressione della malattia
- Ritardo mentale in 1 paziente
- Laboratorio
- CK lievemente elevata
- MRI: Cervello normale
- Patologia
- Variazione nella dimensione delle fibre
- Colorazione ridotta per l'integrina α-7
MD congenita con iperrilassatezza delle articolazioni
63
l
? Integrina-α9 (ITGA9)
; Cromosoma 3p23-21
(3p21.3); Recessiva
- Epidemiologia: Famiglie franco-canadesi del sudovest del Quebec
- Clinica: Simile alla CMD di Ulrich
ma più leggero
- Insorgenza
- Età: Nascita
- Ipotonia con contratture
- Debolezza
- Generalizzata
- Camminare (100%): 1 ÷ 3 anni
- Progressione
- Lento
- Carrozzina 2a ÷ 4a decade
- Respiratorio
- Capacità vitale: Ridotta; Media 50%; Gamma 21%
÷ 100%
- Nessuna progressione
- Nessuna insufficienza
- Articolazioni e Scheletrico
- Contratture: Caviglie (71%), ginocchia (21%), spalle (21%)
- Iperrilassatezza
- Distale + altre
- Dita (93%), polsi (43%), dita dei piedi (43%), gomiti (43%), colonna cervicale
- Scoliosi: Variabile; Da lieve a grave
- Calcagni non protundenti
- Intelligenza: Normale
- Laboratorio
- CK serica: Normale a 1000
- Cardiaco: Normale
- Patologia muscolare
- Dimensione delle fibre: Variazione
- Fibre sottili: Arrotondate o angolari
- Ipertrofia
- Nuclei centrali
- Tessuto connettivo endomisiale: Aumentato
- Predominanza delle fibre muscolari di tipo I
- Vacuoli bordati: Sparpagliati nelle fibre muscolari
- Normali: Distroglicano α; Collagene VI
- Eterozigoti: Iperrilassatezza delle articolazioni
MD congenita con atrofia del CNS e
assenza di grossi assoni mielinizzati nei nervi periferici
l
Recessiva
- Clinica
- Insorgenza: Neonatale
- Artrogriposi multipla congenita
- Ritardo psicomotorio: Grave e generalizzato
- Motorio
- Ipotonia
- Debolezza: Faccia; Assiale e arti
- Caratteristiche dismorfiche
- Decorso: Insufficienza respiratoria e Morte in 1° anno in alcuni
- Patologia
- Ventricolomegalia
- Atrofia: Grave di cervello e cervelletto
MD congenita con atrofia cerebellare
l
? Autosomica recessiva
- Clinica
- Insorgenza: Da neonatale a 7 mesi
- Ipotonia: Generalizzata; Pietre miliari motorie ritardate
- Debolezza: Prossimale > distale
- Cerebellare: Atassia; Nistagmo; Disartria
- Ritardo mentale
- Laboratorio
- CK: 900 ÷ 5.000
- Muscoli
- Variazione nella dimensione delle fibre
- Tessuto connettivo: Aumentato
- Normali distrofina e merosina
- Cerebello: Atrofia; Variante Dandy-Walker
- Materia bianca: Normale
- Non progressiva
Disturbi dei muscoli-occhi-cervello
19
- Alterazioni generali del CNS: Corteccia a "ciottoli"
- Assenza o malformazioni del Gyral
- Corteccia disorganizzata di spessore variabile
- Migrazione neuronale deficiente
- Sindromi di Walker-Warburg (WWS)
- Generale
- Clinica: Sistemi con anomalie
- Disturbi genetici: Generalmente producono una anormale glicosilazione
del distroglicano α
- Altre lissencefalie
- Miller-Dieker
- lissencefalia collegata a X con agenesi del corpo calloso
- Norman-Roberts
- Istologia muscolare
- Miopatia con tessuto connettivo aumentato
- Distroglicano α: Ipoglicosilazione
l
WWS: Mannosiltrasferasi O 1 (POMT1)
; Cromosoma 9q34.1;
Recessiva
- Genetica: POMT1
- Mutazioni
- Missenso: Gly76Arg
- Stop e frameshift: Gln303Stop; Gln385Stop; 2110InsG; 2167InsG
- Trasversione: 1283T to A,
- Eterogeneità genetica
- Le mutazioni POMT1 identificate nel 20% delle famiglie Walker-Warburg: Tipica
sindrome WW
- Allelica con: LGMD con ritardo
mentale e distroglicano α ridotto
- Proteina mannosiltrasferasi O 1 (POMT1)
- Mannosiltrasferasi O
- Isoforma differente dovuta a splicing alternativo
- Espressa ubiquitariamente
- Livelli di picco: Adulti testicoli; Muscoli scheletrici e cardiaco; Cervello fetale
- Funzioni
- Sintetizza il mannosio O collegato al nucleo dell'oligosaccaride con POMgnT1
- POMT1 catalizza il primo passo nella sintesi del mannosilglicano O
- Proteine O mannosilate
- Proteine O mannosilate nei mammiferi: Distroglicano α, N-CAM, tenascina J1
- I glicani O attaccati al distroglicano α
giocano un ruolo nel riconoscimento molecolare della laminina-α2
- La sequenza del mannosilglicano O condivide
- Galβ1-4GlcNAcβ1-2Man-OSer/Thr
- Variante sialata sia nel cervello che nei muscoli:
Neu5Acα2-3Galβ1-4GlcNAcβ1-2Man-O-Ser/Thr
- Localizzazioni della glicosilazione collegata al mannosio O: Cervello, nervi periferici,
glicoproteine muscolari
- Il 30% di tutte le catene di zuccheri collegate ad O nel cervello sono
basate sul mannosio O
- Altri disturbi della glicosilazione: POMGnT1,
CMD di Fukuyama e FKRP,
LARGE
- Epidemiologia
- Distribuzione mondiale della sindrome WW
- Mutazioni POMT1 identificate in famiglie turche ed europee
- Clinica
- CMD più grave con disturbo della glicosilazione
- Motorio: Ipotonia
- CNS
- Attacchi epilettici
- Grave ritardo
- Occhi: Molteplici anomalie anteriori e posteriori; Congenite
- Anteriore
- Cataratta
- Camera anteriore corta
- Microcornea e microftalmia
- Difetti delle lenti
- Posteriore
- Retina: distacco, displasia e neuroni indifferenziati
- Nervi ottici e macula: Ipoplasia e atrofia
- Coloboma
- Altre: Difetti testicolari nei maschi
- Decorso: Morte in feto o nell'infanzia
- Solitamente < 1 anno
- Gamma 0,1 ÷ 3 anni
- Laboratorio
- CK: Variabile; Pazienti con mutazione POMT1 > 1.500
- Patologia muscolare
- Patologia del CNS
- Corteccia
- Difetti migrazionali
- Patologia: Eterotopia neuronale
- Patofisiologia
- Difetti limitanti nella glia piale
- Con
conseguente eccesso di migrazione neurale durante la laminazione della neocorteccia
- Sottile
- Idrocefalo: Ventricoli dilatati
- Lissencefalia: Tipo 2; Corteccia non classica con 4 strati; Ciottoli
- Encefalocele occipitale
- Materia bianca: Mielinizzazione disorganizzata; Corpo calloso assente o sottile
- Cerebello: Afolia; Ipoplasia vermale o generale
- Tronco cerebrale: Piramidi piccole o assenti
- Midollo spinale: Colonne laterali sottili o assenti; Zone di entrata della
radice anteriori aberranti
l
WWS: Mannosiltrasferasi O 2 (POMT2)
; Cromosoma 14q24.3;
Recessiva
- Epidemiologia
- 6 famiglie collegate al locus
- 3 famiglie con mutazioni identificate
- Genetica
- Mutazioni: Omozigotiche R638X; c.1005+1G>A (sito di splicing); T433X
- Proteina POMT2
- Trascritto corto: Ampiamente espresso
- Trascritto lungo: Espresso solamente nei testicoli
- Subcellulare: Associato con reticolo endoplasmatico
- Clinica: Simile ad altre sindromi WWS
- Insorgenza
- Età: Prenatale
- Idrocefalo
- Ipotonia
- Oftalmico: Cataratte; Microftalmo
- Laboratorio
- CK serica: Alta
- MRI: lissencefalia; Idrocefalo; Ipoplasia del ponte e del cervelletto
- Biopsia muscolare
- Dimensione delle fibre: Variata
- Tessuto connettivo endomisiale: Aumentato
- Fibre muscolari basofile
- Colorazione distroglicano α: Ridotta
- POMT2 sindrome variante: LGMD 2N
79
- Epidemiologia: Singolo paziente
- Genetica: Mutazione POMT2
- Omozigotica
- Missenso
- Thr184Met
- Clinica
- Laboratorio
- CK serica: Alta; 1500 ÷ 3300
- EMG: Miopatica
- Biopsia muscolare: Miopatica
- Variazione nella dimensione delle fibre
- Necrosi e rigenerazione
- Cellularità: macrofagi
- Distroglicano α: Ridotto
Distrofia muscolare congenita di Santavuori
(Finnica; Malattia muscoli-occhi-cervello (MEB))
29
l
β1,2-N-acetilglucosaminiltrasferasi collegata al mannosio O (POMGnT1)
; Cromosoma 1p32-p34;
Recessiva
- Mutazioni genetiche POMGnT1
- Tipi: Missenso, nonsenso e frameshift
- Fenotipo più grave con mutazioni verso la 5’ fine del gene
- Funzione della proteina
POMGnT1
- Glicosilazione del mannosile O
- Trasferisce la acetilglucosamina N residua ad un mannosio collegato ad O
- Il mannosile glicano O è necessario per il legame fra il distroglicano α e
la laminina
- ? Ruolo nella migrazione neuronale
- Effetti della mutazione: Funzione enzimatica ridotta
- Epidemiologia
- Focolaio della malattia: Finnici (effetto fondatore)
- Altri: Famiglie turche, francesi, giapponesi e coreane
- Clinica
- Gravità: Più lieve della Walker-Warburg
- Motorio
- Ipotonia: congenita
- Sviluppo: Lento
- CNS: Grave ritardo mentale
- occhio
- Rete trabecolare grossolana nella camera anteriore: Miopia (>-6 diottrie);
Glaucoma
- Displasia retinica
- Cataratta giovanile
- Morte
- Laboratorio
- CK serica: Lieve innalzamento
- Potenziali visivi evocati: Giganti
- Patologia muscolare
- Infanzia: Degenerazione e rigenerazione delle fibre muscolari
- Distrofia: Fanciullezza
- Dimensioni delle fibre muscolari: Variata
- Rigenerazione delle fibre muscolari
- Tessuto connettivo endomisiale: Aumentato
- Componenti del tessuto connettivo
- Patologia del CNS
- Tronco cerebrale: Sottile
- Cerebellare: Ipoplasia con assenza del verme inferiore
- Corteccia: Disorganizzazione (ciottoli); Difetti della linea mediana; Idrocefalo
- Materia bianca: Anomalie, specialmente diffuse nella giovane età < 18 mesi
Distrofia muscolare congenita con epidermolisi bollosa delle articolazioni familiare
l
Plectina (Plec1)
; Cromosoma 8q24.3-qter;
Recessiva
- Genetica: Mutazioni missenso, stop, piccole delezioni e duplicazioni
- Plectina proteina
- Dimensione: 500 kD
- Struttura: Contiene filamenti intermedi e domini leganti actina
- Famiglia: Plachine
- Espressione tissutale: Ubiquitaria; La più alta negli strati squamosi epiteliali,
muscoli e cervello
- Localizzazione subcellulare: Concentrata ai siti dello stress
- NMJ: Membrane postsinaptiche
- muscolo: Sarcolemma e linee Z
- Cute: Emidesmosomi
- Cuore: Dischi intercalati
- Complessata con
- Desmuslina
: Associato con
distrobrevina α
- Desmina
- Altro: Microtubuli, spectrina α, lamina nucleare B, integrina β4, proteine associate ai microtubuli
- Funzioni
- Collegamenti citoscheletrici: Connette
a
- Filamenti intermedi
- Actina
- Microtubuli
- Organelli
- Integrina β4
- Collagene di tipo XVII (BP180)
- Nesprina 3a
- Proteine sarcolemmali
- Integrità meccanica delle cellule: Formazione del citoscheletro di desmina intermiofibrillare
- Substrato per la caspasi 8 durante l'apoptosi mediata da CD95 e dal
recettore del fattore di necrosi tumorale
- Malattia associata: Parziale, ma ridotta, espressione associata con
la malattia più leggera
- Insorgenza
- Congenita
- Debolezza o ipotonia
- Lesioni cutanee: Vesciche
- Clinica
- Cute: Vesciche; Dovute a trauma e calore
- Muscoli
- Debolezza: Miopatia e Sindrome miastenica
- Distribuzione: Prossimale, distale, facciale e oculare
- Decorso: lentamente progressiva; Molti in carrozzina a 10 anni
- Il grado della debolezza può essere correlato con i livelli di espressione della plectina
- Ipotrofia muscolare diffusa
- Sistemico
- Contratture dei gomiti
- Complicazioni respiratorie infantili
- Alopecia
- Denti cariati
- Reti laringee
- Restringimenti uretrali
- Cardiomiopatia, dilatativa, biventricolare
- CNS: Atrofia cerebrale; Ventricoli ingrossati
- Trattamento
- Diaminopiridina 3,4: Qualche miglioramento
- Piridostigmina: Nessun beneficio
- Laboratorio
- CK serica: Alta; > 1.000
- Elettrofisiologia
- Stimolazione nervosa ripetitiva: Decremento
- EMG: Miopatica; Irritabile
- Fisiologia della placca terminale
- Quantitativo rilasciato per impulso: Normale
- MEPP: Piccoli
- AChR: AChR fetali ed adulti alle NMJ
- Patologia muscolare
- Miopatica
- Necrosi e rigenerazione
- Dimensioni delle fibre muscolari: Variata
- Nuclei interni
- Tessuto connettivo: Aumentato
- Anormale architettura interna alla colorazione NADH
- Proiezioni sarcolemmali
simili a punte con colorazione integrina β1D
- Placche terminali: Catene di piccole regioni sulla superficie delle fibre
- Plectina
- Assente o ridotta colorazione nelle linee Z muscolari e cute
- Proteine correlate anormali: BP180; BP230
- Proteina emidesmosomica HD1: Colorazione ridotta
- Desmina: Depositi focali nella maggior parte delle fibre muscolari
- Alcuni pazienti: Bastoncelli;
Corpi citoplasmatici; Vacuoli bordati
- Mitocondri 32
Distrofia muscolare congenita con anormalità strutturali mitocondriali
2
l
Recessiva
- Clinica
- Motorio
- Debolezza: Prossimale; Lentamente progressiva
- Ipotonico; Ritardo nel camminare
- Cardiomiopatia: Dilatativa
- Ritardo mentale
- CK: Leggermente elevata
- Patologia muscolare
- Variazione nella dimensione delle fibre
- Necrosi e rigenerazione
- Mitocondri
- Colorazione SDH e COX
- Aumentata alla periferia delle fibre muscolari di tipo 2
- Ridotta al centro delle fibre (Normale SR in queste regioni)
- EM: Mitocondri ingranditi con struttura anormale
- Merosina: Normale
- Vedi anche: Distrofia muscolare congenita con epidermolisi bollosa alle articolazioni
Distrofia muscolare congenita con iniziale rigidità spinale
Dalla D Cummings MD
|
| MD congenita + spina rigida |
l
Selenoproteina N, 1 (SEPN1)
; Cromosoma 1p35-p36;
Recessiva
- Genetica
- Selenocisteina incorporata nelle proteine al codone UGA
- Il codone UGA è normalmente un codone di stop
- Il riconoscimento di UGA come codone selenocisteina
- Richiede l'inserzione della sequenza (SECIS) in 3'UTR del trascritto
- Secondario all'elemento di ridefinizione (SRE) localizzato adiacente al codone UGA
- Mutazioni SEPN1
- Nonsenso: Frameshift; Codone di stop; Splicing
- Localizzazioni missenso
- Dominio conservato
- Vicino al codone UGA: Elemento SRE
73
- Nella sequenza SECIS
- Le mutazioni causano anche
-
Proteina SEPN1
40
- Famiglia delle selenoproteine: Tutte sono enzimi
- Struttura
- Dimensione: 70 kDa
- Contiene singoli residui di selenocisteina
- Glicoproteina
- Localizzazione subcellulare: Reticolo endoplasmatico; Membrane
- Livelli
- Alti: La maggior parte dei tessuti fetali umani; Cellule proliferanti
- Più bassi nei tessuti adulti
- Muscoli scheletrici: Progressivo decremento come fusi mioblasti
per diventare miotubi
- Funzione: ? Primo sviluppo nelle cellule in proliferazione o rigenerazione
- Selenoproteina
- Catalizza le reazioni di ossidazione e di riduzione
- Contiene
residuo(i) di selenocisteina (Sec) localizzata(e) in un sito enzimatico attivo
- La carenza di selenio: Causa
- Cardiomiopatia (malattia di Keshan)
- Distrofia muscolare nel bestiame
- Epidemiologia
- Famiglie di origine: Marocchina, turca, iraniana, scandinava
- Caratteristiche cliniche: Sovrapponibili alla sindrome della miopatia congenita con mininuclei
-
Debolezza
8:
Gravità variabile
- Insorgenza: Variabile
- Precoce: Nascita ÷ 1 anno
- Tarda: 1a decade
- Insorgenza precoce: Ipotonia; Scarso controllo del capo
- Distribuzione
- Collo; Faccia; Prossimale; Distale
- Simmetrica
- Respiratorio
- Capacità vitale
- Spesso ridotta: Minima al 55% nella 1a decade
- Ipoventilazione notturna: Apnea centrale durante il sonno paradosso
- Insufficienza progressiva
- Camminare: Mai < 18 mesi
- Dimensioni muscolari: Sottili, specialmente nelle coscie interne e
cranici
- Decorso
- Iniziale miglioramento con lo sviluppo
- Poi non progressivo o lento declino
- Scheletro
- Spina
dorsale
- Rigida: Insorgenza 3 ÷ 7 anni; Flessione
limitata del collo e della spina dorsale
- Scoliosi: Insorgenza 4 ÷ 12 anni;
Progressiva
- Altre contratture (50%): Flessori del gomito; Estensori delle anche; Caviglie; Ginocchia
- Cardiaco: Difetti di conduzione minori, o normale
- CNS: Intelligenza normale; Nessuna alterazione della materia bianca
- Endocrino: Insulino-resistenza
- Laboratorio
- CK serica: Normale
- MRI dei muscoli
- Coinvolgimento dei muscoli selettivo
- Perdita di volume e segnale aumentato in T1
- Muscoli coinvolti: Vasto laterale; Bicipite femorale
- Muscoli risparmiati: Vasto mediale; Semitendineo; Adduttore magno;
Gracile
- Patologia muscolare: Scegliere il muscolo per la biopsia con la MRI
- Modello anatomico: Diverso coinvolgimento di muscoli diversi
- Dimensione delle fibre: Variabile
- Fibre muscolari
- Necrosi: Occasionale
- Mininuclei
- Aree di breve lunghezza
- Ultrastruttura: Disorganizzazione sarcomerica e deplezione mitocondriale
- Immunocolorazione: Filamina C; Cristallina αB
- Tipi di fibre: Predominanza delle fibre muscolari di tipo I o sottili;
Presenza di entrambi i tipi
- Tessuto connettivo endomisiale: Aumento da minimo a lieve
- Merosina: Normale
l
Recessiva
- Debolezza: Camminare mai
- Rigidità della spina e scoliosi: Insorgenza molto precoce
Distrofia muscolare congenita con insufficienza respiratoria e ipertrofia muscolare (CMD1B; MDC1B) 7
l
Cromosoma 1q42; Recessiva
- Epidemiologia: Famiglie arabe e tedesche
- Clinica
- Grave coinvolgimento del diaframma: Insufficienza respiratoria
- Debolezza
- Collo
- Prossimale > distale
- Braccia e gambe
- Faccia: Lieve
- Camminare: Insorgenza 18 mesi ÷ 2 anni e mezzo
- Non chiara la progressione della debolezza
- Ipertrofia muscolare: Generalizzata
- Contratture
- Caviglie
- Rigidità spinale (50%)
- Intelletto: Normale
- Laboratorio
- CK serica: Molto alta; 1.700 ÷ 7.600
- Cervello: MRI normale
- Muscoli
- Alterazioni distrofiche
- Proteine del tessuto connettivo: Ridotte in alcune fibre
- Merosina: Ridotta; Nessuna mutazione nel gene merosina
- Integrine α 7 e β1D: Ridotte
- Solfato di eparan proteoglicanico: Normale
Distrofia muscolare congenita con ipertrofia muscolare (MDC1C)
16,26
l
Proteina correlata a Fukutina (FKRP)
; Cromosoma 19q13.3;
Recessiva
- Epidemiologia: Mutazioni in 7 famiglie
- Genetica
- Allelica con
- Mutazioni: Missenso
- Ser221Arg, Ala455Asp, Pro448Leu, Tyr309Cys, Ser385Ter, Pro316Thr,
Tyr465Ser
- La mutazione Tyr465Ser aggiunge mutazione un nuovo sito di potenziale
glicosilazione
- Nessun paziente con 2 alleli FKRP nulli
- I pazienti con la mutazione L276I hanno solitamente la
LGMD 2I
- Proteina
FKRP
- Ubiquitaria
- I livelli più alti nei muscoli scheletrici, cuore e placenta
- Subcellulare: Proteina normale nel Golgi
- Funzione: ? Glicosiltrasferasi o trasferasi fosforil-ligando
- Casi più gravi:
Proteina mutante
54
- Localizzazione: Reticolo endoplasmatico
- Emivita più breve
- Preferenzialmente degradata dai proteasomi
- Clinica
- Insorgenza
- Prima settimana di vita
- Ipotonia
- Ridotti movimento e attività
- Debolezza
- Diffusa: Arti; Collo; Tronco; Faccia
- Abilità presenti
- Può sedere senza assistenza
- Incapacità di camminare
- Dimensioni muscolari
- Ipertrofia: Specialmente polpacci; La maggior parte dei pazienti
- Atrofia: Cingoli delle spalle; Altre muscoli
- Contratture
- Alcuni pazienti
- Ginocchia; Caviglie; Gomiti; Spina
- Scheletro: Capo piccolo; Scoliosi
- CNS
- Solitamente normale
- Occasionali pazienti con
- Lieve ritardo mentale: Psicomotorio; Assenza della parola
- Cisti cerebellari
- Laboratorio
- CK serica
- Molto alta: 3.000 ÷ 8.000
- Si possono avere livelli più bassi con l'aumentare dell'età
- EMG: Miopatica
- Biopsia muscolare
- Dimensione delle fibre variata
- Tessuto connettivo: Aumentato
- Piccole degenerazioni o rigenerazioni
- Laminine
- α2: Parziale riduzione attorno fibre muscolari; Normali nei nervi
- α5: Aumentata
- Distroglicani
- Distroglicano α
- Colorazione muscolare: Marcata riduzione: Alterazioni simili viste nella
CMD di Fukuyama
- Western blot: Anormale peso molecolare
- Distroglicano β: Normale
- MRI cerebrale: Normale o cisti cerebellari
- ECG: Normale
- Vedi anche: CMD con ipertrofia muscolare, ritardo mentale e ipoplasia cerebellare
Distrofia muscolare scleroatonica (Ullrich)
21
l
Collagene, tipo VI, subunità α1 (COL6A1)
; Cromosoma 21q22.3;
Recessiva o dominante
l
Collagene, tipo VI, subunità α2 (COL6A2)
; Cromosoma 21q22.3;
Recessiva o dominante
l
Collagene, tipo VI, subunità α3 (COL6A3)
; Cromosoma 2q37;
Recessiva o dominante
- Genetica
- Mutazioni
- Recessive (COL6A1, COL6A2 e COL6A3)
- Troncamento o missenso
- Ridotto o assente il collagene VI nei muscoli
- Comunemente: Localizzate nel dominio della tripla elica; Alterno ai
residui di glicina
- Dominanti (COL6A1, COL6A2 e COL6A3)
- Delezione o mutazione inframe
-
Effetto negativo dominante
41: Interferisce con
la formazione del dimero e la secrezione della
molecola
- Spesso nella regione terminale N: Prossimale alla cisteina nel dominio della tripla elica
- Storia familiare della malattia insolita
- Allelica con
- Alcuni pazienti con sindrome simile: Non collegata a questo loci;
Collagene VI
normale
- Proteina collagene VI
- Proteina ubiquitaria della matrice extracellulare
- Concentrata sul lato esterno della lamina basale
- Muscoli e fibroblasti nella miopatia di Ullrich
- Mal localizzata, scarsa o assente
- Caratteristiche cliniche
- Variabili
- Alcuni con grave debolezza
- Alcuni famiglie con la mutazione COL6A3 hanno una malattia più leggera
- Insorgenza
- I movimenti fetali possono essere ridotti
- Contratture congenite
- Ipotonia
- Articolazioni
- Inizialmente
- Contratture
- Prossimali > distali
- Le contratture alle ginocchia possono limitare il camminare
- Spina dorsale: Rigidità; Cifoscoliosi
- Torcicollo
- Può migliorare con l'aumentare dell'età
- Debolezza
Dalla: A Connolly MD
Ipercheratosi pilari
|
- Diffusa: Distale > prossimale; Flessori del collo
- Deambulazione
- Possono camminare 1 ÷ 2 anni (30%)
- La maggioranza non cammina mai
- Insufficienza respiratoria
- Insorgenza: Ipoventilazione nella 1a decade
- Insufficienza: Non correlata con il grado della debolezza
- Atrofia muscolare: Predominanza distale
- Decorso
- Lentamente progressiva
- Camminare
- Alcuni camminano per un tempo limitato: 3 ÷ 6 anni
- Morte
- Età: 1a o 2a decade
- Associata con l'insufficienza respiratoria
- Cute
- Morbida
- Cheratosi pilare
- Cheloidi
- Cicatrici atrofiche
- Smagliature
- Petecchia
- Scheletro
- Calcagni prominenti
- Scoliosi
- Spina rigida
- Tendini: Simili alla Ehlers-Danlos; Contratture
- Cardiaco: Normale
- Intelligenza: Normale
- Caratteristiche di laboratorio
- CK serica: Normale ÷
elevata10x
- EMG: Miopatica
- Biopsia muscolare
- Dimensioni variate delle fibre muscolari: Alcune fibre muscolari molto sottili
- Tessuto connettivo endomisiale: Progressivo aumento
- Fibre muscolari necrotiche: Rare o occasionali
- Fibre Lobate: Occasionali
- Espressione del collagene
- Tipo VI
- 2 modelli della patologia
- Assente dai muscoli scheletrici e dai capillari
- Assenti sulla superficie delle fibre muscolari ma presente nel tessuto connettivo
- Nessuna correlazione tra il modello della patologia ed il fenotipo clinico
- Tipo IV: Sovraregolata
- Ultrastruttura
- Membrana plasmatica muscolare: Irregolari, estensione e ripiegamento
- Lamina basale: Ispessimento e sovrapproduzione; Replicazione della
membrana basale dei capillari
- Morfologia dei capillari 42:
lumi piccoli; Aumento delle vescicole pinocitotiche nelle cellule endoteliali
- Collagene: Fibrille disorganizzate
- Esame genetico: Utah
Distrofia muscolare congenita con grave ritardo mentale e
glicosilazione anormale (MDC1D) 43
l
LARGE
; Cromosoma 22q12.3-13.1;
Recessiva
- Epidemiologia: 1 paziente
- Genetica
- Quinto gene umano per grandezza
- Mutazioni
- Missenso: Glu509Lys
- Inserzione (1bp): 1999insT
- Altre mutazioni nel: Topo myd
- Proteina
LARGE
- Espressa ubiquitariamente
- I livelli più alti in cuore, cervello e muscoli scheletrici
- Due domini catalitici putativi con omologia alle
- Glicosiltrasferasi α (famiglia batterica WaaIJ)
- Sintesi dei lipopolisaccaridi della membrana esterna
- Acetilglucosaminiltrasferasi β 1,3 N (iGnT)
- Sintesi acetillattosamina poi N spina dorsale dell'antigene degli eritrociti i
- Può avere un ruolo nella glicosilazione del
distroglicano α 47
- Insorgenza
- Età: 5 mesi
- Ritardato nello sviluppo globale
- Clinica
- Distrofia muscolare congenita
- Ipotonia
- Lento miglioramento nella 1a decade
- ipertrofia muscolare
- Moderata
- Quadricipite; Polpacci; Muscoli delle braccia
- Lingua: Normale
- Atrofia muscolare: Nessuna
- Debolezza
- Prossimale > distale
- Gambe > braccia
- Faccia: Debolezza lieve
- EOM: Normale
- CNS
- Ritardo mentale: Profondo
- Movimenti specchiati
- Nistagmo: Laterale
- Spasticità: Tono aumentato; Riflessi tendinei aumentati; Estensore
plantare
- Contratture
- Laboratorio
- CK serica: Alta; 400 ÷ 4.500
- NCV: Normali motorio e sensorio
- EMG: Miopatica
- Elettroretinogramma: Risposte evocate piccole
- Imaging del CNS
- Alterazioni della materia bianca: Dalle fibre periventricolari alle
arcuate; Anteriori e temporali
- Anormalità strutturali: Migrazione neuronale anormale; Tronco cerebrale sottile
- Patologia: Muscoli
- Distroglicano α
- Immunomarcatura: Ridotta
- Immunoblotting con epitopi glicosilati: Ridotto peso molecolare del distroglicano α
- Conservate alcune attività leganti della laminina
- Laminina α2: Normale
- Disferlina: Sovraregolata nel Topo myd
- Modello di topo: Muscoli del topo myd
CMD con pollici addotti, oftalmoplegia e ritardo mentale
34
l
Autosomica recessiva
- Epidemiologia: Fratelli e sorelle siciliani consanguinei
- Insorgenza: Congenita
- Clinica
- Articolazioni: Pollici addotti; Contrattura delle dita dei piedi; Iperlordosi
- Motorio: Ipotonia; Ritardo nel camminare
- Debolezza: Diffusa; Facciale; Grave
- Oculare: Oftalmoplegia; Ptosi
- Cognitivo: Ritardo mentale, IQ 65 ÷ 70
- Laboratorio
- CK serica: Alta, 130 ÷ 750
- NCV: Normale
- MRI: Lieve ipoplasia cerebellare
- Muscoli
- Distrofici: Dimensione delle fibre variata; Aumento del tessuto connettivo endomisiale
- Normale laminine e distroglicano α
Distrofia muscolare congenita con ipertrofia muscolare, microcefalia e
ritardo mentale 15
l
Autosomica recessiva
- Epidemiologia: Bambini italiani
- Insorgenza
- Clinica
- Ipotonia
- Contrattura delle articolazioni: Anche;
Ginocchia; Caviglie: Gomiti; Flessori del collo
- CNS: Ritardo psicomotorio; Assenza della parola
- Motorio: Incapacità a camminare; Debolezza diffusa
- Ipertrofia muscolare: Polpacci; Quadricipite; Lingua
- Oculare: Normale
- Decorso: Non progressivo
- Laboratorio
- CK serica: Molto alta
- Biopsia muscolare
- Alterazioni distrofiche
- Fibre muscolari sottili
- Tessuto connettivo aumentato
- Piccole degenerazioni o rigenerazioni
- Laminine
- α2: Parziale riduzione
- α5: Aumentata
- MRI cerebrale
- Fossa posteriore: Cisterna magna ingrandita; Verme cerebellare ±
ipoplasia degli emisferi
- Materia bianca: Segnale a chiazze, specialmente periventricolare
- ECG: Normale
- Vedi anche: CMD con cisti cerebellari
Distrofia muscolare congenita 55
l
Cromosoma 4p16.3; Recessiva
- Epidemiologia: Famiglia palestinese, consanguinei
- Insorgenza
- Età: Neonatale o in utero
- Movimenti fetali ridotti
- Ipotonia
- Clinica
- Motorio
- Iniziale: Ipotonia; Ridotta movimenti anti gravità
- Debolezza
- Prossimale > distale
- Più gravi: Muscoli dei cingoli del tronco e della spalle
- Coinvolgimento da lieve a moderato: Faccia, collo, prossimali degli arti
- Nessuna: Extra-oculare, Bulbare
- Dimensioni muscolari: Sottili, non ipertrofici
- Decorso
- Alcuni muoiono nell'infanzia o adulti: 50% con infezioni respiratorie
- Motorio: Statico
- La maggior parte dei sopravissuti capaci di camminare da bambini e da adulti
- Articolazioni
- Non contratture
- Lieve lassità negli adulti
- Intelligenza: Normale
- Laboratorio
- CK serica: Normale o minimamente elevata
- CT cerebrale: Normale
- Ecocardiogramma: Normale
- Patologia muscolare
- Morfologia: "Distrofica"
- Normale colorazione: Laminine (α2, α5, β1, β2), Distrofina, Sarcoglicani
(α, β, γ), α e Distroglicano β
ALTRE DEBOLEZZE CONGENITE
Distrofia miotonica congenita
l
Proteina miotonina chinasi; Cromosoma 19; Dominante
- La più grande # ripetizione di triplette di qualsiasi sindrome MD (> 1.000)
- Esaminare genitori, specialmente la madre, per i segni della malattia
Distrofia facioscapoloomerale (FSH) congenita
l
Cromosoma 4q35; Dominante
- La FSH congenita spesso si presenta senza che i genitori siano
chiaramente affetti
Miopatie metaboliche
congenita Sindromi miasteniche
Sindromi neuropatiche congenite
Escludere l'ipotonia CNS
Malattia della banda A larga
- Epidemiologia: 2 casi sporadici
- Clinica
- Insorgenza: Neonatale
- Ipotonia
- Ritardo nello sviluppo: Motorio; Linguaggio
- Progressivo miglioramento delle funzioni
- Distrofia retinica: Un paziente
- Laboratorio
- CK serica: Leggermente alta
- EMG: Normale
- Patologia muscolare
- Bande A larghe visibili nelle sezioni plastiche
- Disallineamento dei filamenti spessi della miosina
Miopatia trilaminare
- Epidemiologia: 1 caso sporadico
- Clinica
- Insorgenza: Nascita
- Tone: Aumentata resistenza ai movimenti passivi
- Contratture
- Decorso: Aumento dei movimenti spontanei
- Cognizione: Normale
- Laboratorio
- CK serica: Alta (6x ÷ 40x)
- EMG: Normale
- Patologia muscolare
- Dimensione delle fibre: variata
- Fibre grosse: Apparenza trilaminare
- NADH: Zone scure interne ed esterne
- ATPasi: Zone pallide interne ed esterne
- Ultrastruttura
- Zone centrali mancanti di miofibrille organizzate
- AChR: Extragiunzionali, tra zone intermedie ed esterne
Miopatia con corpi zebrati
l
Actina α (ACTA1; Muscoli scheletrici)
; Cromosoma 1q42.1;
Sporadica
- Epidemiologia: 2 casi sporadici; ? entità cliniche
- Mutazioni: Alleli nulli
- Clinica
- Insorgenza: Nascita
- Ipotonia
- Debolezza
- Simmetrica o asimmetrica
- Può essere maggiore nelle braccia
- Dimensioni muscolari: Piccole
- Riflessi tendinei: Normali
- Decorso: Statico o lento miglioramento
- Laboratorio
- CK serica: Normale o leggermente alta
- EMG: Miopatica
- Patologia muscolare
- Dimensione delle fibre: variata
- Corpi zebrati
- Materiale delle linee Z in pile separate da filamenti sottili
- Possono trovarsi nei muscoli normali
- Bastoncelli nemalinici
Debolezza con precoci disturbi scheletrici
-
Sindrome campodattilia di Tel Hashomer
l
Autosomica recessiva
- Caratteristiche cliniche
- Scheletrico: Campodattilia (dita flesse); Club piedi; Spina bifida -
Cervicale
- Cute: Assente crescita interfalangea; Verticilli di 7 o più dita; Crescita
palmare
- Pori sudoriferi anormali
- Cardiaco: Prolasso della valvola mitrale
- Miopatia: Atrofia; Fibre di dimensioni variabili; CK serica alta
- Eterozigoti: Parziali manifestazioni scheletriche (campodattilia del
5o dito) e
cutanee
- Sindrome di Camera-Marugo-Cohen
- Caratteristiche cliniche
- Obesità
- Ritardo mentale
- Scheletrico: Asimmetria corporea; Campodattilia; Clinodattilia
- Debolezza muscolare: Prossimale, gambe > braccia 50%; Ipotonia congenita 50%
Sindrome Williams-Beuren
l
Sindrome del gene contiguo autosomica dominante; Cromosoma 7q11.23
l
Geni cancellati: Elastina
; RFC2
; Chinasi LIM
; Fattore di trascrizione muscolare
GTF3 (MusTRD1); GTF2I
- Genetica
- Microdelezione in regioni di DNA altamente ripetitivo
- 2° a ricombinazione fra sequenze ripetute disallineate fiancheggianti
regioni WBS
- Clinica
- Stenosi vascolare: Aortica sopravalvolare; Coronaria; Polmonare; Renale
- GU: Stenosi ureterale; Reni piccoli o solitario
- ENT: Faccia da elfo; Voce rauca;
Paralisi delle corde vocali
- Gastrointestinale: Ernia inguinale; Costipazione; Diverticolosi
- Metaboliche: Disidratazione episodica; Ipertensione; Ipercalcemia
- Ritardo mentale: 65 IQ medio
- Abilità visivo-spaziali carenti
- Relativamente preservata la funzione verbale
- Personalità "Cocktail party"
- Deficit di attenzione
- Ipersensibilità al suono
- Contrattura delle articolazioni
- Motorio
- Infanti: Ipotonia; Ritardo nel camminare; Affaticamento
- Adulti: Crampi; Mialgia; Affaticamento
- Patologia muscolare
- Miopatia: Fibre di dimensioni variabili
- Alcuni con accumulo lipidico e carenza di carnitina
Miopatia perifascicolare neonatale 1
- Insorgenza
- Età: Neonatale
- Motorio: Ipotonia e debolezza
- Clinica
- Motorio
- Ipotonia
- Debolezza: Diffusa
- CNS
- Ritardo cognitivo
- Ritardo mentale: Lieve
- Decorso
- Non progressivo
- Miglioramento nelle abilità motorie con l'aumentare dell'età
- Patologia muscolare
- Fibre muscolari: Modello
perifascicolare della patologia
- Atrofia
- Fibre muscolari immature, tipo 2C
- Tessuto connettivo
- Colorazione fosfatasi alcalina del perimisio
- Cellule: macrofagi o dendritiche
- Infiammazione: Perivascolare, v vicino e immediatamente attorno ai vasi
locali
Disturbo immunoneurologico (Sindrome Woods-Black-Norbury)
l
Dominante; Cromosoma Xq26-qter
- Maschi: Ipotonia neonatale; Morte precoce
- Femmine: Paraparesi spastica
- Debolezza: Progressiva
- Riflessi tendinei: Vivaci
- Disfunzioni della vescica
- Visione notturna: Ridotta
- Carenza di IgG2
Assenza congenita, debolezza, o ipoplasia muscolare
Muscoli extraoculari
- Fibrosi congenita dei muscoli extraoculari (CFEOM)
l
CFEOM1
KIF21A (cinesina)
; Cromosoma 12p11.2-q12;
Dominante e sporadica
- Mutazioni 46
- Eterozigotiche
- Missenso: R954W; R954Q; Altre
- Regione gambo della proteina: La più comune
- Alcune de novo: Più nei casi sporadici
- Può anche produrre il fenotipo CFEOM3
- Proteina
KIF21A
- Proteina motoria
- Classe delle cinesine: La stessa delle KIF21B e KIF4
- Altre mutazioni della cinesina
- Clinica
- Ptosi: Congenita; Bilaterale
- EOM
- Difetto comune: Incapacità ad alzare gli occhi
- Altro: Movimenti orizzontali ridotti
- Occhi fissati 20° ÷ 30° sotto l'orizzontale
- Capo pendente: Posteriore
- Blefarofimosi
- Patologia
- Assenza di sviluppo della divisione superiore del nervo oculomotorio
- Ridotti # i neuroni oculomotori del mesencefalo α
- Anormali inserzioni muscolari extraoculari
- Alterazioni muscolari: Retto superiore e elevatore delle palpebre superiori
- Vedi anche variante clinica: FEOM 3A
l
CFEOM2
ARIX (PHOX2A)
; Cromosoma
11q13.3-q13.4; Recessiva
- Mutazioni: Siti di splicing e missenso
- Proteina ARIX
- Fattore di trascrizione dell'omeodominio
- Necessario per lo sviluppo dei nervi cranici III e IV
- Epidemiologia: Arabia Saudita (Ala72Val); Turchia
- Clinica
- Movimenti extraoculari limitati
- Occhi fissati in abduzione
- Carente funzione dei muscoli innervati dal terzo o dal quarto nervo cranico
- Gli occhi possono alzarsi al di sopra dell'orizzontalità o avere un coinvolgimento unilaterale
- Ptosi
- Acuità visiva
- Scarsa
- Grave ambliopia: In occhi con pupille coperte da palpebre ptotiche
- Pupille
- Miotiche
- Fissate o reattive lentamente alla luce o ai dilatatori pupillari
l
CFEOM3
Tubulina, Beta-3 (TUBB3)
; Cromosoma 16q24.3;
Dominante
- Genetica
- Mutazioni TUBB3
- Missenso
- R62Q; Arg262Cys (Comune); R262H; Ala302Thr; R380C; Glu410Lys; Asp417His;
Asp417Asn
- Localizzazioni: Regioni della tubulina β regolanti le dinamiche dei microtubuli,
traffici ed interazioni della proteina motoria con le MAP
- E410, D417 e R262: Perdita di cinesina localizzata alle finale plus dei
microtubuli
- A302T, R62Q e R380C: Microtubuli più stabili e maggiore aspettativa di vita
- R262C, R262H e E410K: Durata prolungata degli eventi di crescita dell'individuo;
Aumento del tasso di depolimerizzazione
- Simile alla sindrome CFEOM3 vista con: Alcune mutazioni KIF21A
- Proteina TUBB3
- Può avere un ruolo nella guida alla crescita e nel mantenimento degli
assoni
- Le mutazioni possono danneggiare la formazione dell'eterodimero della
tubulina
- Può interagire con KIF21A
- Clinica
- Espressione variabile
- Spesso asimmetrica
- Ptosi
- Movimenti extraoculari: Limitati
- Funzione carente dei muscoli innervati dal terzo o dal quarto dei nervi cranici
- Gli occhi possono alzarsi al di sopra dell'orizzontale o avere un coinvolgimento unilaterale
- Lievi: Sguardo verticale limitato
- Grave: 1° sguardo fissato in posizione ipo- ed esotropica; Marcata restrizione
degli EOM
- Marcus-Gunn: Elevazione delle palpebre ptotiche associata con
movimenti sincinetici della mascella
- Altre caratteristiche: Alcune mutazioni
- Aberrante innervazione della muscolatura craniale da parte del nervo trigemino
- Insufficienza intellettuale e comportamentale : A302T, E410K, R262H e R380C
- Paralisi facciale
- Polineuropatia assonale sensoria e motoria: A tarda insorgenza; E410K
D417H o mutazioni R262H
- Età dell'insorgenza: 1° ÷ 3a decade
- Debolezza delle gambe
- Perdita sensoria
- EMG: Cronica, polineuropatia sensoria e motoria generalizzata
- Può avvenire senza CFEOM in alcuni pazienti: Sindrome simile alla
CMT 2
- Contratture dei polsi e delle dita: Mutazioni D417H o R262H
- Decorso: Non progressivo
- MRI
- Ipoplasia: Nervo oculomotorio
- Ipoplasia: Muscoli innervati dal III nervo
- Divisione superiore: Elevatore delle palpebre superiori; Retto superiore
- Divisione inferiore: Retto mediale
- Innervazione aberrante del nervo oculomotorio: Muscolo retto laterale
- Disgenesi del corpo calloso, commissura anteriore (AC) e capsula interna
- Più gravi: A302T, E410K, R262H e R380C
- Meno gravi: R62Q, R262C, o D417N
- Materia bianca: Perdita generalizzata
- Gangli basali: Dismorfismi correlanti con specifiche mutazioni
- Corteccia: Normale
l
CFEOM con
anomalie ulnari nella mano 58
Cromosoma 21qter; Recessiva
- Epidemiologia: 1 famiglia turca
- Clinica
- Oculare
- Oftalmoplegia
- Muscoli
- Specialmente inferiore obliquo e retto superiore
- Sguardo verso l'alto limitato: La più comune
- Elevatori delle palpebre: Alcuni pazienti
- Casi gravi: Oftalmoplegia completa
- Non progressiva
- Duzioni forzate: Normali
- Blefaroptosi: Occhio destro
- Postura del capo: Mento innalzato
- Patologia EOM: Fibrosi
- Mani
- Oligodattilia/oligosindattilia postassiali
- Specialmente assenza del 5° dito
- Più grave nel lato destro
Sindrome di Duane

Clicca sull'immagine per il movimento
|
|
Sindrome di Duane
|
- Caratteristiche cliniche: Generali
- Restrizione della abduzione, della adduzione, o di entrambe
- Cause della adduzione degli occhi
- Retrazione dei globi
- Restringimento delle fessure palpebrali
- Strabismo: Esotropia la più comune
- Solitamente unilaterale e sporadica
- Sindromi bilaterali
- Ambliopia e strabismo: Le più comuni
- Specialmente nei casi familiari
- Associazione con malformazioni congenite comuni
- Scheletriche
- Orecchie
- Oculari
- Nervose
- Patologia
- Assenza dei nuclei e dei nervi abducenti
- Muscolo retto laterale
- Denervato
- Aberrante innervazione della divisione inferiore del nervo oculomotorio
(Anche muscolo retto mediale innervato omolateralmente)
- DURS1
l
Cromosoma 8q13; ? Sporadica
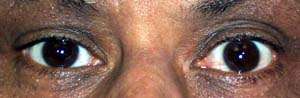
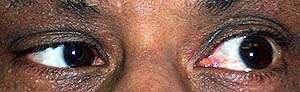
|
Fenomeno simil-Duane in paziente
diagnosticato come miastenia grave oculare cronica
- Lievi ptosi nell'occhio destro con sguardo centrato
(sopra)
-
Ptosi aumentata e retrazione nell'occhio destro
con adduzione (sguardo a sinistra). (sotto)
|
- Genetica: Delezione nella regione di riarrangiamento 8q
- Clinica: Disturbo multisistemico
- Debolezza della abduzione
- Retrazione dell'occhio e ptosi con adduzione
- Associata alla fusione vertebrale C2-C3 (Klippel-Feil)
- Ipoplasia tenare
- Sordità
- DURS2
72
l
α2-Chimerin (CHN1)
; Cromosoma 2q31;
Dominante
- Genetica: Mutazioni
- Missenso: Eterozigotiche
- Aumento della funzione: Attività aumenta della chimaerina α2 RacGAP
- Aumento di alcune mutazioni
- Traslocazione della chimaerina α2 alla membrana cellulare
- Abilità della chimaerina α2 ad auto-associarsi
- Effetto funzionale: disturbo assonale
- Aberranti ramificazioni e/o defascicolazioni del nervo
- Proteina CHN1
- Rac specifiche attivanti GTPasi: Abbassanti l'attività Rac
- Lega al diacilglicerolo (DAG), un estere del
forbolo segnalante lipidi associato alla membrana
- L'espressione più forte durante lo sviluppo nel mesencefalo e rombencefalo
- Oftalmoplegia congenita
- Adduzione: Assente o ridotta
- Adduzione: Ridotta in alcuni pazienti
- Retrazione dell'occhio e ptosi nella adduzione
- Incidenza più alta di
- Coinvolgimento bilaterale
- Anormalità nei movimenti verticali
- Patologia
- Assenza del nervo abducente e del nucleo nel ponte
- Nervi oculomotori ipoplastici: Muscoli innervati dall'oculomotorio
piccoli
- Topo con perdita di chimaerina α2
- Distrutta la segnalazione efrina/EphA4
- Elevati livelli di RacGTP
- Fenotipo: Andatura simile al coniglio saltellante risultante da
- Patologia: Eccessiva e aberrante linea mediana e degli assoni
attraversanti il tratto corticospinale
e proiezioni degli interneuroni spinali
- Anomalia di Duane + anormalità dei raggi radiali e sordità (DRRS;
Sindrome di Okihiro)
l
Sal-like 4 (SALL4)
; Cromosoma
20q13.13-q13.2; Dominante
- Allelica con: Sindrome IVIC
- Clinica
- Arti superiori: Ipoplasia del muscolo eminenza tenar; Assenza del
pollice e del primo metacarpale
- Oculare: Sindrome di Duane; Coloboma
- Renale: Malrotazione; Assenza
- Orecchie: Sordità; Rimpicciolimento esterno e del meato uditivo
- Diagnosi differenziale: Sindrome di Holt-Oram
- Sindrome di Bosley-Salih-Alorainey (BSAS)
l
Omeobox A1 (HOXA1)
; Cromosoma 7p15.3;
Recessiva
- Epidemiologia: Famiglie arabo-saudite, turche e native americane
(Navajo)
- Genetica
- Mutazioni
- Stop: 175-176insG, R26X, Y28X
- Omozigotiche
- Le mutazioni in altri geni HOX: Causano malformazioni delle estremità distali
- Clinica
- Sindrome di Duane: Bilaterale
- Sguardo orizzontale: Adduzione e adduzione limitate
- Sguardo verso alto e verso il basso: Preservato
- Convergenza: Normale; Associata a retrazione dei globi
- Sordità e mutismo (89%)
- Sviluppo motorio (66%): Ritardato; Camminare > 3 anni
- Altro sul CNS (22%)
- Ritardo mentale
- Disturbo dello spettro dell'autismo
- Malformazioni
- Carotide
- Ipoplasia o assenza dei canali e arterie craniali: Carotide interna
spesso assente
- Ingrandimento compensatorio delle arterie comunicanti basilari
e posteriori
- Orecchie: Assenza della coclea, dei canali semicircolari e del vestibolo
- VI nervo: Assente
- Normali: Cerebello, tronco cerebrale, cerebro
- Disturbo allelico: Athabascan (Navajo) sindrome da disgenesi del tronco cerebrale (ABDS)
- Mutazione: R26X
- Insorgenza: Congenita
- Clinica: BSAS +
- Debolezza facciale
- Ipoventilazione centrale
- Paralisi delle corde vocali: 20%
- CNS
- Ipoventilazione
- Ritardo nello sviluppo: Globale
- Attacchi epilettici: 50%
- Cuore
- Difetti congeniti: 50%
- Anomalie del tratto defluente in alcuni
- Esami di BAER e calorico: Nessuna risposta
- Sindrome simile al topo HOXA1-/-
- Altre Duane multisistemiche o sporadiche
- Alterazioni cariotipiche nei cromosomi 4 o 22
- Sindrome di Wildervanck
- Anomalia di Duane
- Anomalia di Klippel-Feil (vertebre cervicali fuse)
- Sordità percettiva congenita
- Vista principalmente nelle femmine
- Sindrome di Bosley-Salih-Alorainey (BSAS)
Sindrome Möbius
PEO (paralisi dello sguardo orizzontale) e scoliosi
l
ROBO3
; Cromosoma 11q23-25;
Recessiva
- Epidemiologia: 10 famiglie, turche, indiane, pachistane, greche, arabe
- Insorgenza: 1a decade o congenita
- Mutazioni: Missenso; Inserzione di 1 bp; Sito di splicing
- Proteina ROBO3
- Espressa negli assoni commissurali nel tronco cerebrale
- ? Permette agli assoni di attraversare la linea mediana
- Clinica
- Paresi dello sguardo
- Orizzontale: Nessun movimento orizzontale liscio coordinato, saccadi, o
riflesso vestibolari
- Verticale: risparmiato
- ? Sopranucleari
- Esotropia: 30%
- Nistagmo: Pendolare
- Miochimia facciale (1 paziente)
- Scoliosi: Da lieve a moderata
- Ritardo mentale: 30%
- Nessuna debolezza
- MRI
- Tronco cerebrale: Displastico in alcuni pazienti
Muscolo superiore retto
- Collegamento esterno:
DJO
Anomalia di Axenfeld-Rieger
l
Dominante
- Occhi
- Iris anteriore ipoplastica
- Assenza parziale di muscolatura oculare
- Proptosi
- Ipertelorismo
- Orecchie: Sordità neurosensoria, lieve
- CNS
- Idrocefalo comunicante
- Ritardo psicomotorio
- Bassa statura
Ptosi congenita, ereditaria
l
Tipo 1
; Cromosoma 1p32-p34.1;
Dominante
- Ptosi: Simmetrica o asimmetrica
- Movimenti oculari normali
- Penetranza: Incompleta
l
Tipo 2
; Cromosoma Xq24-q27.1;
Dominante
- Maschi = Femmine
- Penetranza: Alta
- Insorgenza: Congenita
- Bilaterale: Simmetrica
- Movimenti oculari normali
Sindrome di ROCA
Ritardo nella crescita e nello sviluppo
Oculare: Ptosi
Cardiaco: Difetto
Atresia anale
- Clinica
- Ritardati: Crescita; Mentale
- Faccia: Ptosi, epicanto, ponte nasale largo, orecchie situate in basso e anormali
- Difetto cardiaco congenito
- Atresia anale
Blefarofimosi: Fessura palpebrale ridotta nella dimensione orizzontale
- Blefarofimosi 1
: + Ptosi e epicanto
inverso
l
FOXL2
; Cromosoma 3q23;
Dominante
- Genetica
- Espansioni polialaniniche ripetute (30%): Possono causare una sindrome blefarofimosica
senza insufficienza ovarica
- Clinica
- Ptosi
- Telecanto
- Fimosi
delle palpebre
- epicanto inverso: Piccole pieghe cutanee crescono dalla palpebra inferiore
e corrono su e giù in direzione del naso
- ± Insufficienza ovarica
- Sindrome di Saethre-Chotzen (SCS;
Blefarofimosi 2)
l
TWIST
Cromosoma 7p21; Dominante
- Blefarofimosi con bassa statura
l
Autosomica recessiva
- Famiglia: Yemenita
- Clinica
- Occhi: Blefarofimosi; Ptosi; Oftalmoplegia
- Faccia: Prognatismo; Sinofirie; Sopracciglia spesse; Fronte sfuggente
- Scheletro: Bassa statura; Sindattilia delle dita dei piedi 2 e 3
- CNS: Ritardo mentale borderline; Anosmia
- Sindrome di Ohdo
- Ritardo mentale
- Malattia cardiaca congenita
- Occhi: Blefarofimosi; Blefaroptosi
- Denti: Ipoplastici
- Sindrome di Schwartz-Jampel
- Sindrome di Dubowitz
- Ptosi
- Blefarofimosi
- Telecanto laterale
- Scheletro
- Microcefalia
- Bassa statura
- Ritardo mentale (50%)
- Sindrome di Marden-Walker
Faccia
Depressor anguli oris: Sindrome cardiofacciale
l
Cromosoma 22q11.2 (delezione) Dominante; Anche multifattoriale
- Nosologia: Sindrome di Cayler; Sindrome di Di George
- Genetica: Spesso derivata da cromosomi paterni
- Debolezza facciale: Gravità variabile
- Congenita
- Lieve: Labbro più basso da un lato
- Grave: Paresi facciale completa
- Cardiaco
- Difetto del setto atriale
- Difetto del setto ventricolare
- Miopatia
20
- Forza: Normale
- CK serica: Può essere alta
- Biopsia muscolare: Variazione nella dimensione delle fibre; Glicogeno aumentato
- Scheletro
- Fusione vertebrale cervicale
- Pollici addotti
- Dismorfismo facciale
- Ugola bifida
- Immunitario
- Cellule T anormali
- Ipoplasia timica
- CNS (10%)
- Microcefalia: Variabile
- Ritardo mentale
Vedi anche: Paresi facciale congenita
Assenza dei muscoli della masticazione: Modello di topo
- Doppio knockout per la MyoR (Musculina)
e capsulina (TCF21)
- Fenotipo
- Assenza dei muscoli in 1° arco branchiale: Massetere; Pterigoidi, mediale e
laterale; Temporale
- Palatoschisi
- Ernia diaframmatica: Posteriore
Debolezza del diaframma 78
- Nervo frenico: Generale
- Muscoli principale dell'inspirazione
- Innervazione motoria
- Radici: C3, C4, C5
- Nervo frenico
- Innervazione sensoria
- Nervo frenico
- Nervi intercostali
- Lesioni
- Gli infanti: Clinicamente affetti più gravemente
- Clinica: Unilaterale
- 50% Asintomatica
- 25% dispnea da esercizio, lieve
- Capacità vitale: 75% del previsto
- Clinica: Bilaterale
- Dispnea da esercizio: Grave
- Ortopnea
- Ipossiemia e ipercapnia notturne
- Capacità vitale: 45% del previsto; Supina < 75% della posizione
eretta
- Radiologia: Diaframma elevato nel lato affetto
- Ereditaria e congenita
e collegata a X
- Generale
- Riportate
bilaterale e lato sinistro
- Solitamente recessiva
- Tipi generali di ernia diaframmatica congenita (CDH)
- Bochdalek
- 70 ÷ 90% dei casi di CDH
- Lateralizzata a sinistra
- Posterolaterale
- Morgagni: Anteriore retrosternale
- CDH centrale
- Linea mediale del setto trasverso
- 1 ÷ 2% delle CDH
- Sindromi specifiche
- Ernia diaframmatica
- DIH1
: Cromosoma 15q26.1
- DIH2
: Cromosoma 8p23.1
- Frequentemente unilaterale
- Associazioni: Ipoplasia del polmone sinistro; Difetto del setto atriale
- DIH3
: Proteina dito a zinco,
multitipo 2 (ZFPM2)
; Cromosoma 8q23
- Difetti diaframmatici con ossificazione del cranio e carenze negli arti
: ? Autosomica recessiva
- Ernia diaframmatica anteriore congenita
- ? collegata a X vs. multifactoriale con alto rapporto maschi:femmine
- Morte neonatale frequente
- Acquisite
- Traumatiche
- Eziologie
- Intervento chirurgico
- Intervento chirurgico a cuore aperto
- Trapianto polmonare
- Intervento chirurgico esofageo
- Procedure mediastinali
- Trauma al collo o al torace
- Associazione con lesioni al
plesso brachiale
- Decorso: Peggioramento bifasico
- 1° ore: Tachipnea e necessità di O2
- Giorni ÷ settimane: Atelettasia o infezione
- Diagnosi: Fluoroscopia del torace
- Prognosi
- Recupero: 50%; Insorgenza in 1° o 2° settimane
- Mortalità: 10% per unilaterale e 50% per bilaterale
- Trattamento: Plicazione del diaframma affetto
- Lesioni del nervo frenico: Altro
- Neoplasmia
- Herpes zoster 77
- SLA
- Disturbi muscolari
- Malattia polmonare ostruttiva cronica (COPD)
- Fibre muscolari atrofiche
- Alterazioni delle fibre: Più nel tipo I; Perdita di miosina; Distruzione del sarcomere
- Amiloide
- Vedi anche: Insufficienza respiratoria
Mani
Sindrome di
Holt-Oram
l
Fattore di trascrizione umano TBX5
; Cromosoma 12q24.1;
Dominante
- Cardiaco: ASD; VSD; AV blocco
- Scheletro: Bilaterale; Sinistra > Destra nel 70%
- Pollice (85%): Assenza; Ipoplasia; Trifalangeo
- Radio (64%): Assenza o ipoplasia
- Contratture: Gomito; Polsi
- Casi gravi: Focomelia
- Muscoli assenti o ipoplastici
- Eminenza tenar (100%): Pollice non opponibile
- Trapezio: Scapole alate; Spalle spioventi
- Altre: Deltoide; Bicipiti; Tricipiti; Estensori dei polsi; Abduttori ulnari; Supinatore
- Arti inferiori: Normali
Muscolo palmaris longus
l
Autosomica dominante con penetranza incompleta
- Più frequente: Femmine; Lato sinistro
Estensori delle dita e dei pollici
l
Autosomica recessiva
- Ipoplasia unilaterale dell'estensore del dito
- Contratture flessorie
- Polineuropatia: Sensoria con ipoidrosi
- Vedi anche: Holt-Oram
Tronco
Pettorali (Sindrome di Poland)
l
Solitamente sporadica
- Generale
- Maschi: Femmine = 2,4:1
- Destra: Sinistra = 1,7:1
- Prevalenza: 1 su 30.000
- Insorgenza: ? 6a settimana di gestazione
- Manifestazioni cliniche: Variabili
- Aplasia muscolare
- Capo sternale del pettorale
- Altro: Serrato anteriore; Latissimo dorsale; Glutei; Infraspinato e sopraspinato
- unilaterale: La maggior parte
- Anomalie della mano: omolaterali, ipoplastiche
- Sindattilia semplice
- Dita corte
-
Anomalie più gravi
28
- Collegamento esterno:
Radiologia
- Anomalie craniofacciali
- Sindrome di Möbius
- Ptosi
- Prognatismo mandibolare craniofacciale
- Displasia craniofrontonasale
- Sindrome di Romberg
- Anomalie dell'orecchio esterno
- Anomalie scheletriche
- Spinale
- Toracica
- Costole, clavicole e sterno
- Deformità di Sprengel
- Petto scavato e carenato
- Arti inferiori: Reti poplitee; Piede a clava; Sindattilia delle dita
- GI: GI ernia; Coliti ulceranti; Stenosi pilorica; Erniazione epatica
- GU: Agenesi renale; Riflusso ureterale; Testicoli non discesi
- Caratteristiche sistemiche: Altre
- Peli ascellari: Assenza a zone
- Aplasia di mammella e capezzolo
- Cardiovascolare
- Destrocardia
- Difetto del setto atriale
- Malformazioni vascolari
- Neoplasmie: Occasionali
- Teratoma toracico
- Fibroma pleurico
- Ematopoietiche: Leucemia; Linfoma
- Vedi anche
Trapezio
l
Cromosoma 8q12.2-q21.2; Dominante
- Gene contiguo alle sindromi
- Vedi anche: Holt-Oram
Legamento scapolare traverso superiore: Calcificazioni
l
Autosomica dominante
Muscolatura addominale: Sindrome Prune belly
l
Autosomica; ? Recessiva, dominante o sporadica
- Nosologia: Prune belly; Sindrome Eagle-Barrett
- Piena sindrome più comune nei maschi
- Clinica
- Assenza dei muscoli addominali: Distensione addominale
- Ostruzione urinaria; Criptorchidismo
- Ano imperforato
- Scheletro: Petto scavato; Piede a clava; Lussazione delle anche
- Cardiaco: Dotto arterioso pervio
Gambe
Muscolo
peroneo terzo
l
Autosomica recessiva
-
Muscolo
peroneo terzo
- Presente negli umani e negli equini
- Innervazione: Nervo peroneo profondo
- Azione: Dorsiflessione ed eversione dei piedi durante la fase ondeggiata
dell'andatura
- Livella il piede ed aiuta le dita dei piedi a non toccare il suolo
- Migliora
l'economy dei bipedi nel camminare
- Assenza dei muscoli
- Frequenza: 4,4% ÷ 15% degli arti
- Più frequente nel lato destro (14:9)
Psoas (Sindrome CHILD)
l
Proteina steroidea simile alla deidrogenasi
NAD(P)H
; Cromosoma Xq28;
Dominante
l
Proteina legante a emopamile
; Cromosoma Xq28;
Dominante
- Emidisplasia congenita con eritroderma ittiosiforme e difetti
negli arti - Congenital Hemidysplasia con Ichthyosiform
erythroderma e Limb Difetti
- Ipoplasia
- Sistemico: Polmoni; Tiroide
- Psoas muscolare
- Nervi cranici V, VII, VIII, IX e X
- CNS: Ponte; Midollo; Cervelletto; Midollo spinale
Malattia dei multinuclei (mininuclei)
12
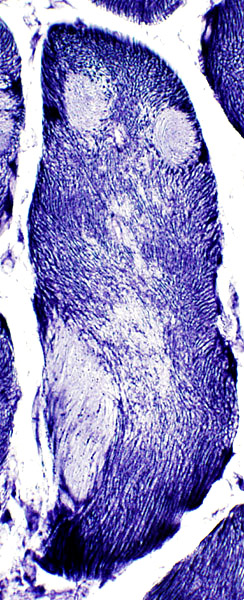
|
| Multinuclei |
- Genetica: Numerose associazioni
l
Recettore rianodina
; Cromosoma 19q13.1;
Recessiva
l
Selenoproteina N, 1 (SEPN1)
; Cromosoma 1p35-p36;
Recessiva o sporadica
l
Carenza di acil–CoA deidrogenasi a catena corta
l
Titina
l
Recessiva o sporadica; ? Famiglie con dominante
- 50% delle malattie multinuclei non collegate a RYR1 o SEPN1
- Insorgenza
- Età: Prenatale alla fanciullezza
- Pietre miliari motorie ritardate
- Clinica: Caratteristiche generali
- Quattro presentazioni generali
- Caratteristiche solitamente normali
- Livelli della creatina chinasi serica
- Intelletto
- Cardiaco
- Non ipertermia maligna
- Clinica: Forma comune; Più grave
- Genetica: Comuni mutazioni SEPN1
- Caratteristiche della insorgenza precoce
- Movimenti fetali: Ridotti
- Ipotonia
- Debolezza
- Inizialmente
- Ritardo motorio
- Infine tutti in grado di camminare
- Distribuzione
- Assiale > prossimale > distale
- Estremità inferiori e superiori
- Facciale: Comune; Lieve
- Oculare
- Ptosi
- Oftalmoplegia
- Alcuni pazienti
- Paresi da parziale a completa (10%)
- Respiratorio: Più comune con
- Età > 10 anni
- Scoliosi > 60°
- Ipoventilazione notturna
- Decorso: Non o lentamente progressivo
- Scheletro
- Contratture: Lievi o assenti
- Lassità dei ligamenti
- Piedi: Piatti o estroversi; Ginocchio valgo
- Ginocchia: Iperestendenti
- Lordosi lombare
- Spina rigida: Lievi; 25%
- Scoliosi: > 10 anni
- Cardiaco (Occasionali): Difetti del setto; Blocco di conduzione; Cardiomiopatia
- Intelligenza: Normale
- Non ipertermia maligna
- Laboratorio
- CK: Normale o leggermente elevata (20%)
- EMG: Miopatica, specialmente > 4 anni e muscoli prossimali
- MRI muscolare: Coinvolgimento del vasto; Retto del femore risparmiato
- Patologia: Muscoli
- Dimensioni delle fibre muscolari: Variabile
- Caratteristiche distrofiche: Alcuni pazienti
- Fibre muscolari rigeneranti
- Tessuto connettivo endomisiale: Aumentato
- Molti piccoli cori nelle singole fibre muscolari
- Istochimica: Ridotta attività alle colorazioni NADH e mitocondriale
- Brevi: Non si estendono per l'intera lunghezza delle fibre muscolari
- Frequenza: Presenti nel 60% ÷ 90% delle fibre muscolari
- Si ritrovano in entrambi i tipi I e II delle fibre muscolari
- Più pronunciati con il crescere dell'età
- Ultrastruttura: Regioni centrali non strutturate; Sarcomere alterato
- Fibre muscolari di tipo I
- Nuclei interni
- Multinuclei: Varianti genetiche
- Gruppo di mutazioni selenoproteina N, 1 (SEPN1)
- Ereditarietà: Recessiva o sporadica
- Allelica con: MD congenita con spina rigida
- Frequenza: 30% delle famiglie con mini multinuclei
- Clinica: Sovrapposta alla MD congenita con spina rigida
- Generale: Malattia a mininuclei "tipica" più grave
- Motorio
- Debolezza assiale
- Insufficienza respiratoria
- Preservate: Forza negli arti; Deambulazione
- Massa muscolare: Amiotrofia
- Scheletro: Spina rigida; Scoliosi
- Muscoli: Nuclei centrali più piccoli che con la mutazione RYR1
- Malattia a mininuclei con oftalmoplegia
- Genetica: Numerosi geni associati
- RYR1: Dominante o recessiva
- SEPN1
- Fenotipo riportato con la carenza di acil–CoA deidrogenasi a catena corta
- Insorgenza: Congenita o 1° anno
- Motorio
- Ipotonia
- Difficoltà di alimentazione
- Coinvolgimento assiale e del cingolo della spalla
- Funzione respiratoria: Ridotta; Occasionali insufficienze
- MRI muscolare: Maggior coinvolgimento del sartorio e del soleo
- Peso: Ridotto
- Scoliosi
-
Artrogriposi ad insorgenza prenatale
33
- Artrogriposi, generalizzata
- Capo
- Dolicocefalia
- Radice nasale prominente
- Fessure palpebrali oblique
- Palato ad arco alto
- Orecchie più in basso
- Collo breve: Con lievi pterigio colli
- Spina: Cifosi o cifoscoliosi, precoce e grave
- Altre caratteristiche
- Torace a campana
- Clinodattilia,
- Criptorchidismo, bilaterale
- Piega palmare singola
- Debolezza
- Prossimale
- Lieve
- Capacità vitale ridotta
- Fenotipo lieve della sindrome multinuclei: "Simile alla malattia del nucleo centrale"
- Epidemiologia: Quattro famiglie
- Genetica
- Ereditarietà recessiva
- Mutazioni del recettore rianodina (O collegate a 19q31.1)
- Mutazioni RYR1: Omozigotiche; Missenso; P3527S
- Sovrapposta a fenotipo della sindrome del nucleo centrale:
Missenso V4849I
- Clinica: Fenotipo lieve
- Insorgenza: Infanzia o fanciullezza
- Debolezza
- Gravità: Moderata
- Distribuzione: Prossimale, cingolo pelvico e assiale; Mani
- Respiratorio
- Capacità vitale variabilmente ridotta: 50%
÷ 100% del normale
- Nessuna insufficienza respiratoria
- Altri muscoli
- Massa preservata
- Dolore allo sforzo
- Articolazioni
- Iperrilassatezza
- Lussazione delle anche
- Artrogriposi
- Patologia muscolare
- Alterazioni miopatiche
- Mininuclei o nuclei centrali: Più grandi che con la mutazione SEPN1
- Colorazione ATPasi: Tutte le fibre muscolari di tipo I
- Miopatia congenita + cardiomiopatia dilatativa fatale: Recessiva
- Epidemiologia: Famiglie marocchine e sudanesi
- Mutazioni Titina
- Delezioni omozigotiche
- Fuori frame
- Clinica
- Pietre miliari motorie ritardate
- Ipotonia alla nascita: Alcuni pazienti
- Debolezza
- Insorgenza: < 1 anno
- Simmetrica
- Generalizzata: Prossimale + distale + faccia
- Gambe > braccia
- Ptosi: Alcuni asimmetrica
- Decorso: lentamente progressivo; Deambulazione presente nella 2a decade
- Dimensioni muscolari
- Arti superiori: Atrofia
- Gambe: Pseudoipertrofia, specialmente coscie e polpacci
- Contratture
- Rigidità spinale
- Articolazioni e collo
- Cardiomiopatia
- Progressiva
- Dilatativa
- Età dell'insorgenza: 5 ÷ 12 anni
- Disturbi del ritmo: Ventricolare
- Fibrosi interstiziale
- Morte improvvisa
- CK serica: Leggermente alta
- Muscoli
- Distruzione del complesso proteico della linea M sarcomerica: Le titine
troncate mancanti del Terminale C incorporate dentro i sarcomeri
- Mininuclei: Orli irregolari
- Nuclei centrali
- Miopatia: Aumento del tessuto connettivo endomisiale; Più tardi nell'infanzia
- Calpaina-3 ridotta
- Allelica con
- Portatori della mutazione: Normali
Miopatia congenita con eccesso
di filamenti sottili
5
l
Actina α (ACTA1; Muscoli scheletrici)
; Cromosoma 1q42.1;
Sporadica
- Mutazioni del gene actina
- Gly15Arg: Residui esposti sulla superficie
- Val163Leu: Nucleo idrofobo
- Clinica
- Ipotonia
- Insufficienza respiratoria
- Cardiomiopatia
- Decorso: Variabili con la stessa mutazione actina
- Morte nel 1° mese: 2 pazienti
- Sopravvivenza all'infanzia: 1 paziente
- CK serica: Normale
- Muscoli
- Masse dei filamenti sottili (Actina)
- Nucleare ± bastoncelli
citoplasmatici
- Variazione nella dimensione delle fibre
- Altre sindromi actina α
- Miopatia con bastoncelli
Sindrome di Carey-Fineman-Ziter
l
Autosomica recessiva
- Insorgenza: Congenita
- Clinica
- Miopatia
- Non progressiva
- Ipotonia
- Ipoplasia muscolare
- Muscoli pettorali e omerali (Poland)
- Spesso bilaterale
- Sindrome di Möbius
- Oculare
- Oftalmoplegia
- Ptosi
- Fessure palpebrali: Inclinate verso il basso
- VII nervo: Debolezza facciale, bilaterale
- X nervo
- Disfagia
- Disturbi dell'alimentazione (100%)
- Craniofacciale
- Macrocefalia
- Naso triangolare
- Sequenza di Robin
- Micrognatismo
- Glossoptosi: Lingua situata più indietro; Può ostruire le vie respiratorie
- Palatoschisi (50%)
- Scheletro
- Scoliosi (40%)
- Talismo (40%)
- Equinovaro
- Brachidattilia (70%)
- Ritardo nello sviluppo
- CNS: Intelligenza solitamente normale
- Laboratorio
- Elettrofisiologia
- NCV: Normale
- EMG: Unità motorie piccole; Fascicolazioni
- CK serica: Normale
- Biopsia muscolare
- Variazione nella dimensione delle fibre
- Degenerazione
- Rigenerazione (Desmina + fibre)
- Tipi di fibre: Predominanza del tipo II; Tipo I piccole
Miopatia con riduzione di corpi 3,4
l
Proteina LIM quattro e mezzo1 (FHL1)
; Cromosoma Xq26-q27.2;
Dominante o semidominante
- Genetica
- Mutazioni: Missenso; H123Y, H123Q, C132F, C153R, C153Y
- Localizzazione generale: Residui coordinanti zinco nel secondo dominio LIM
- 123 Mutazioni istidina: Spesso nuove
- 153 residui di cisteina coordinanti zinco: Fenotipo più lieve
- Allelica con
- Proteina FHL1
- Localizzazione cellulare
- Mioblasti: Nucleare
- Miotubi differenziati: Citosolica
- Effetti della mutazione
- Induce la formazione di inclusioni simili ad aggresomi
- Le inclusioni incorporano sia FHL1 mutante che FHL1 di tipo selvatico
- Altre proteine intrappolate in una maniera negativamente dominante
- Sindromi cliniche 75
- Grave: Infantile e fanciullezza
- Storia familiare: Spesso sporadica
- Insorgenza
- 1 ÷ 4 anni
- Più grave: Ritardo motorio nel 1° anno
- Non congenita
- Debolezza
- Prossimale > distale
- Progressione: Rapida; Può avvenire dopo una fase stabile
- Respiratoria
- Scheletro
- Spina rigida: Alcuni pazienti
- Scoliosi
- Contratture: Variabili
- Successivamente nel decorso della malattia
- Disfagia
- Faccia: Ptosi
- Esito fatale: < 10 anni in alcuni pazienti
- A tarda insorgenza: Adulti
6
- Età dell'insorgenza: Fino alla 4a decade
- Clinica
- Sviluppo iniziale: Normale
- Debolezza
- Prossimale > distale
- Scapoloperonea
- Asimmetrica
- Insufficienza respiratoria
- Muscoli paraspinali: Possono essere coinvolti
- Coinvolgimento da progressivo a diffuso
- Maggior prominenza che nei genitori
- Scheletro: Meno che nella forma ad insorgenza infantile
- Spina rigida: Alcuni pazienti
- Scoliosi
- Cardiomiopatia: Dilatativa; Alcuni pazienti
- Femmine portatrici
- Possono essere sintomatiche
- Più segni con distorsioni nella inattivazione di X
- Laboratorio
- CK serica: Aumentata a 3x il normale
- EMG
- Modelli misti neurogenici e miogenici
- Può mostrare attività spontanea
- Miopatia scapoloperonea (Corpi ialinici),
di tipo I
- Miopatia congenita: Insolita
- Ritardato nello sviluppo delle pietre miliari
- Decorso benigno
- ? Differente disturbo
- Biopsia muscolare
- Dimensioni delle fibre muscolari: Variabili
- Attività della fosfatasi acida aumentata
- Corpi riducenti
- Localizzazione: Citoplasmatica; Spesso vicino nucleo
- Colorazione istochimica
- Tricromica
- Inclusioni verdi scure
- ± Associazione con corpi citoplasmatici eosinofili
- NBT menadione (senza glicerofosfato α): Positivo
- Meccanismo: Riduce il tetrazolio blu nitro (NBT)
- Contiene proteine simili ad aggresomi
- Desmina
- Ubiquitina
- Chaperone endoplasmatico del reticolo luminale GRP78
- Tubulina y
- FHL1
- MYBPC1
- Pericentrina
- Può essere più comune nelle fibre muscolari di tipo I
- Vacuoli bordati in alcuni pazienti
- Ultrastruttura
- Corpi riducenti
- Struttura: Materiale granulare tubulo-filamentoso (17 nm in diametro)
- Localizzazione: Periferia del sarcoplasma e attorno al nucleo
- Corpi citoplasmatici: Comuni
- Immunocitochimica: Corpi riducenti
- Contengono proteine normalmente associate con nucleoli
- Colorazione anche per distrofina, sarcoglicano α, vimentina e ubiquitina
- Altri disturbi con corpi riducenti
Miopatia con corpi ad impronta digitale
- Epidemiologia: Qualche caso sporadico
- Ereditarietà: Sporadica o recessiva
- Insorgenza: Congenita
- Clinica
- Debolezza
- Prossimale > distale: Arti e tronco
- Simmetrica
- Risparmiati i nervi cranici
- Ipotonia
- Scompenso intellettivo: Occasionale
- Decorso: Non progressivo o lentamente progressivo
- Laboratorio
- CK serica: Alta in alcuni pazienti
- EMG: Miopatica
- Biopsia muscolare: Inclusioni
- Istochimica: Ovoidi; 1 ÷ 10 μM; Rossi
a H e E; Verdi a GT
- Ultrastruttura: Serie di lamelle osmofiliche parallele ricordanti una impronta digitale
- Corpi ad impronta digitale visibili anche nelle
Miopatia congenita con
alterazioni apoptotiche 9
- Epidemiologia: Identificato 1 paziente giapponese sporadico
- Clinica
- Insorgenza: Ipotonia congenita
- Sviluppo psicomotorio: Marcatamente ritardato
- Motorio
- Ipotonia
- Atrofia muscolare: Generalizzata
- Faccia miopatica
- Riflessi tendinei: Normali
- Contratture: Caviglie
- Laboratorio
- CK serica: Normale
- Siero Lattato: Normale
- Patologia muscolare
- Fibre muscolari
- Sottili
- Basofile: Sparse
- Positive alla fosfatasi acida: Molte
- Immature (Tipo 2C): 40%
- Vacuoli: 20% delle fibre
- Fibrosi endomisiale: Lieve
- Nuclei: Condensazione di cromatina; Frammentazione
- Alterazioni correlate all'apoptosi
- Fibre muscolari positive a bax (Pro-apoptotiche): 80%
- Fibre muscolari
non positive a Bcl-2 (Anti-apoptotiche)
- Caspasi-3 attivata nelle
fibre: 6%
- Nuclei positivi a TUNEL: 2%
Miopatia con corpi ialini
Miopatia con corpi ialini
44
l
Miosina - Catena pesante β cardiaca (MYH7)
; Cromosoma 14q11.2-q13;
Sporadica o dominante
- Nosologia: Chiamata anche
- Miopatia con lisi delle miofibrille di tipo I
- Miopatia con accumulo di miosina
- Genetica
- Mutazione
- Missenso
- Arg1845Trp; His1904Leu
- Localizzazione
- Regione a bastoncelli della MiHC lenta/cardiaca β
- Esoni 37 ÷ 39
- Eterozigotica
- Altre causate
da mutazioni MYH7
- Caratteristiche cliniche
- Penetranza variabile
- Insorgenza: Infanzia o nella prima fanciullezza
- Motorio
- Inizialmente
- Ipotonia
- Pietre miliari motorie ritardate
- Debolezza
- Prossimale ± Distale
- Scapoloperonea
- Braccia e gambe
- Simmetrica
- Può essere grave e disabilitante
- Decorso: lentamente o non progressivo
- Sistemico: Occasionali aritmie cardiache
- Laboratorio
- CK serica: Normale o leggermente elevata
- EMG: Miopatica
- Biopsia muscolare
- Corpi ialini
- Subsarcolemmali
- Eosinofili
- Maggiormente nelle fibre di tipo I
- Mancanza dell'attività ossidativa o glicogena
- Può colorare per ATPasi miosinica (Acido pH 4,3)
- Ultrastruttura
- Matrice di materiale granulare e filamentoso
- Può essere subsarcolemmale
- Non legami di membrana
- Alcune inclusioni intranucleari
- Predominanza delle fibre muscolari di tipo I
- Corpi ialini notati anche in una
miopatia scapoloomerale ad insorgenza adulta
Miopatia con corpi ialini: Scapoloperonea Tipo
1 ad insorgenza adulta
l
FHL1; Cromosoma Xq26; Dominante
Miopatia con corpi ialini: Scapoloperonea Tipo
2 ad insorgenza adulta
l
Autosomica dominante
Miopatia con corpi ialini:
Scapoloperonea Tipo 3
l
Cromosoma 3p22.2–p21.32; Recessiva
Miopatia congenita con eccesso di fusi muscolari
18
- Epidemiologia: Singolo caso; Non storia familiare
- Clinica
- Insorgenza
- Nascita
- Ipotonia
- Artrogriposi
- Debolezza
- Diffusa
- Insufficienza respiratoria
- Ariflessia
- Faccia: Incavamento frontale; Guance flaccide; Bocca triangolare; Palato ad arco alto
- Artrogriposi
- Cardiomiopatia: Tachicardia atriale;
Insufficienza cardiaca; Cardiomiopatia ipertrofica
- Splancnomegalia
- Neuroblastoma: Congenito; Adrenale
- Decorso: Morte a 14 mesi
- Laboratorio
- Studi metabolici normali
- MRI cerebrale:Allargati spazio subarachnoide e ventricolo
- EMG, RNS e NCV: Normali
- Patologia muscolare
- Fibre muscolari atrofiche sparse
- Fusi muscolari: Numero
eccessivo, 1 o più per fascicolo, specialmente nel deltoide e nei
quadricipiti
- Altri disturbi con anormalità dei fusi muscolari
- Distrofia miotonica: Numero eccessivo di
fibre muscolari intrafusi per fuso
- Sindrome di Noonan: Numero aumentato di fusi
- Sovra-espressione di NT 3 (Topo):
Numero eccessivo di fusi
Miopatia congenita con Caps
57
Miopatia Cap: Mutazioni TPM2
l
Tropomiosina β 2 (TPM2)
; Cromosoma 9p13.2-p13.1;
Dominante
- Epidemiologia: Riportati ~5 casi sporadici
- Genetica: TPM2
- Mutazione: Delezione inframe; Glu139del; Eterozigotica
- Allelica con
- Mutazioni non presenti in: Pazienti con miopatia Cap recessiva
- Proteina TPM2
- Clinica
- Insorgenza: Congenita o fanciullezza
- Motorio
- Ipotonia
- Debolezza
- Prossimale e distale
- Faccia
- Respiratoria
- Insufficienza respiratoria : Casi gravi
- Decorso
- Debolezza lentamente progressiva
- Malattia grave: Morte a 14 anni in 1 paziente
- Scheletro
- Faccia: Lunga e stretta
- Palato: Ad arco alto
- Torace: Deformità; Scoliosi
- Laboratorio
- CK serica: Leggermente elevata o normale
- EMG: Miopatica
- Patologia muscolare
- Caps
- Colorazione: Scure alla tricromica di Gomori
- Più comuni nelle in fibre muscolari di tipo I
- Regioni alla periferia delle fibre muscolari
- Colorazione per: NADH; Esterasi; Desmina; Actinina α; Tropomiosina; Actina;
SERCA2; Nebulina; Miotilina
- Ultrastruttura
- Materiale della linea Z anormale
- Architettura miofibrillare disorganizzata
- Fibre muscolari di tipo I: Sottili
Sindrome
del ritardo mentale collegata a X (Cabezas)
14
l
Cullina 4B (CUL4B)
; Cromosoma Xq23;
Recessiva
- Genetica: Mutazioni missenso e stop
- Proteina CUL 4B
- Ubiquitina ligasi
- Complessi con recettore della diossina (AHR)
- Insorgenza
- Nascita ÷ fanciullezza
- CNS: Peso ridotto; Pietre miliari ritardate
- Scheletro: Bassa statura; Piedi piccoli; Spazio tra 1° e 2° dito dei
piedi; Articolazioni iperestensibili; Cifosi
- Obesità
- Testicoli piccoli
- Labbro inferiore prominente
- Neurologico
- Tremore: Fine; Arti superiori
- Coordinazione motoria fine: Ridotta
- Ritardo mentale
- Comportamento: Durata dell'attenzione ridotta; Iperattività; Balzi d'umore;
Aggressività
- Motorio: Andatura a gambe larghe; Necessità della carrozzina
- Atrofia muscolare: Nella parte inferiore delle gambe
Debolezza congenita con diarrea e sordità
l
Recessiva
- Clinica
- Debolezza
- Ipotonia alla nascita: Difficoltà ad alimentarsi
- Faccia miopatica
- Riflessi tendinei: Ridotti
- Miglioramento con l'età
- Contrattura delle articolazioni
- Diarrea: Episodica con infezioni intercorrenti; Secretorio
- Cute: Eruzioni bollose; Si risolvono in 2 mesi
- Sordità: Neurosensoria
- Microcefalia
- Criptorchidismo: Nei maschi
- Laboratorio
- Patologia muscolare: Fibre muscolari sottili, sparse, spesso di tipo II
- Associate alla diarrea: Carenza di zinco; Disturbi degli elettroliti
- Fosfatasi alcalina: Bassa
Sproporzione congenita delle dimensioni fra tipi di fibre
51
- Definizione
- Insorgenza
- Età
- Congenita o nella fanciullezza
- Asintomatica (4%)
- Debolezza
- Biopsia muscolare patologia: Dimensione bimodale delle fibre
- Fibre muscolari di tipo 1 di diametro 12% più piccolo del tipo 2
- Clinica: Caratteristiche generali
- Storia familiare: 43%; Dominante o recessiva
- Motorio
- Debolezza
- Estremità (95%): Diffusa o prossimale > distale
- Grado: Da lieve a grave (27%)
- Faccia (40%)
- Bulbare (35%): Associata con debolezza più grave
- Respiratoria (28%)
- Ipotonia
- Riflessi tendinei: Ridotti o assenti
- Oculare
- Oftalmoplegia (19%): Prognosi peggiore
- Intelligenza: Normale
- Scheletrico (25%)
- Decorso: Statica (55%); Miglioramento (36%); Lenta progressione (9%)
- Laboratorio
- CK serica: Normale o < 3x il limite superiore del normale
- EMG: Miopatica o alcune caratteristiche neuropatiche
- Patologia muscolare
- Variazione nella dimensione delle fibre
- Grado: Media = Fibre di tipo 1 di diametro 41% più piccolo del tipo 2; Gamma fino a 74%
- Correlazione: Più grandi dimensioni disparità con più grave caratteristiche cliniche
- Fibre di tipo 2B < 5% o assenti: 35%
- Predominanza delle fibre di tipo 1 (50%)
- Nuclei interni o centrali (> 5%): 22%
- Bastoncelli nemalini (6%)
- CFTD: Sindromi genetiche specifiche
l
Actina α (ACTA1)
; Cromosoma 1q42.1;
Dominante
- Mutazioni
- Eterozigotica
- Missenso: Leu221Pro; Asp292Val; Pro332Ser
- Differenti dalle mutazioni della miopatia con bastoncelli
- Localizzazione: L'actina superficiale è rimossa dalla tropomiosina
durante l'attività muscolare
- Frequenza nella CFTD: Insolita; 6%
- Clinica: Fenotipo grave
- Insorgenza: Congenita; Debolezza
- Debolezza: Diffusa; Grave; Respiratoria
- Non oftalmoplegia
- Decorso: Insufficienza respiratoria; Morte < 4 anni (2 dei 3 pazienti
- Patologia muscolare
- Fibre muscolari di tipo 1: Sottili; 45% ÷ 54%
del tipo 2
- Non bastoncelli
- Altri disturbi dell'actina α
l
Selenoproteina N, 1 (SEPN1)
; Cromosoma 1p35-p36;
Recessiva 61
- Genetica
- Clinica
- Sindrome simile alla SEPN1
- Ipotonia troncale: Precoce
- Forza
- Collo debolezza: Precoce
- Deterioramento respiratorio: Adolescenza alla prima età adulta
- Forza negli arti: Relativamente preservata
- Scoliosi
- Progressiva
- Lieve dalla fanciullezza all'adolescenza
- Lordosi correlata alla spina superiore
- Insulino-resistenza
- Istologia muscolare
- Fibre muscolari di tipo 1 mediamente di diametro 12% più piccolo del tipo 2
- Non lesioni multimininuclei
l
CFTD2
; Cromosoma Xq13.1-q22.1;
Recessiva
- Epidemiologia: Famiglia australiana
- Insorgenza
- Età: Congenita
- Ipotonia e debolezza
- Clinica
- Debolezza
- Ptosi: Bilaterale
- Faccia
- Suzione e pianto: Pianto debole
- Ipotonia: Generalizzata
- Insufficienza respiratoria
- Cardiomiopatia: Dilatativa; In un sopravissuto
- Decorso
- Morte nella maggior parte: Insufficienza respiratoria; Età 6
÷ 14 settimane
- Sopravviventi: Debolezza facciale e lieve generalizzata
- Laboratorio
- CK serica: Normale
- Lattato: Normale
- Biopsia muscolare: Fibre muscolari di tipo 1 assottigliate
- Portatori
- Debolezza lieve: Generale e faccia (Frontale)
Ritorna a
Indice delle miopatie e giunzioni neuromuscolari
Ritorna a
Neuromuscolare Home Page
Riferimenti
1. Pediatric Neurology 1996;15:150-152
2. Muscle & Nerve 1998;21:40-47
3. J Neurol Sci 1995:128:58-65
4. Neuromuscular Disorders 1998;8:162-168
5. Neuromuscular Disorders 1997;7:160-168
6. Neuromuscular Disorders 1999;9:580-586
7. Am J Hum Genet 2000 February 66:
8. Ann Neurol 2000;47:152-161
9. Ann Neurol 2000;47:531-536
10. Human Mutation 2000;15:393-409, Hum Mutat 2002;19:114-121
11. Human Mutation 2000;15:410-417
12. Neuromuscular Disorders 2000;10:264-273, 2004;14:754–766
13. Am J Hum Genet 2000;67:000
14. J Med Genet 2000;37:663-668
15. Neuromuscular Disorders 2000;10:541-547
16. Neuromuscular Disorders 2000;10:548-552; 2003;13:gennaio online
17. Hum Mol Genet 2000;9:3083-3090
18. Muscle & Nerve 2001;24:138-143
19. Neurology 2001;56:1059-1069
20. Neuropediatrics 2001;32:107-109
21. PNAS 2001;98:7516-7521, Ann Neurol 2001;50, Am J Hum Genet 2002;70:1446-1458
22. Ann Neurol 2001;50:, Trends Mol Med 2001;7:362-368
23. J Neuropath Exp Neurol 1982;41;298-314
24. Neuropath Appl Neurobiol 1994;20;232-237
25. Neuromuscular Disorders 2001;11:538-541
26. Neuromuscular Disorders 2001;11:635
27. Neuromuscular Disorders 2002;12:13-18, cervello 2003;126 Online June
28. Br J Plast Surg 2001;54:132-136
29. Developmental Cell 2001;1:717–724
30. Ann Neurol 2002;51:750-759
31. Neuromuscular Disorders 2002;12:392–398
32. J Neuropath Exp Neurol 2002;61:520-530
33. Ann Neurol 2000;48:745-757
34. Neuromusc Disord 2002;12:623-630
35. Neurology 2002;59:613-617
36. J Neurol Sci 2002; Online ottobre,
J Neuropathol Exp Neurol 2007;66:57-65
37. PNAS 2002;99:15060-15065
38. Neuromuscular Disorders 2002;12:novembre online
39. Neurology 2003;60:1363–1365
40. Human Molecular Genetics 2003;12:1045–1053
41. Am J Hum Genet 2003; Online luglio, Umani Molecolare Genetiche 2005;14:279–293
42. Acta Neuropathol 2003;106:137–142
43. Human Molecular Genetics 2003; Online settembre
44. Ann Neurol 2003;54:494–500
45. Brain 2003;126:2341-2349
46. Nature Genet 2003;Online novembre
47. Nature Medicine 2004;Online June
48. Curr Opin Neurol 2004;17:205–209
49. Neurology
2004;62:1484–1490
50. Hum Genet 2003;113:297–306
51. J Neuropath Exp Neurol 2003;62:977–989, Ann Neurol 2004; Online ottobre
52. Neuromuscular Disorders 2004;14:785–790
53. Neuromuscular Disorders 2004;14:779–784
54. Umani Molecolare Genetiche 2005;14:295–305
55. Umani Genetiche 2005; Online maggio
56. Neurology 2005;64:1638–1640
57. J Neurol Sci 2002;201:27–31,
Disturbi neuromuscolari 2007; Online aprile
58. J Med Genet 2005;42;408-415
59.
Neurology 2005;65: Online settembre 7
60.
Nature Genetics 2005; Online ottobre 16
61.
Ann Neurol 2005; Online Dec
62.
Neuromuscular Disorders 2006; Online Jan
63. Brain 2006; On-line June
64. Neuromuscular Disorders 2006;16:S62
65. Am J Hum Genet 2006; Online ottobre
66.
Ann Neurol 2006; Online dicembre 22
67.
Neuromuscul Disord 2007 maggio 28
68.
Nature Genetics 2007; Online agosto
69. Neurology
2008;70:114–122
70. Ann Neurol 2008; Online Feb
71. Ann Neurol
2007;62:666-670
72. Science 2008 Jul 24
73. Human Mut 2008; Online
Dec
74. Neuromuscular Disorders 2009; Online gennaio
75. Brain 2009 Jan 29
76. Prenat Diagn 2009 Mar
5
77. Muscle Nerve
2009;38:1070-1073
78. Semin Respir Crit Care
Med 2009;30:315–320
79. Biochemical
Biophysical Research Communications 2007;363:1033–1037
80. Neurology
2009;73:1159-1161
81. Pflugers Arch 2010;
Online Feb
82. Neuromuscular
Disorders 2010; Online Mar
27 marzo 2010
Va a Acronimi e sigle
Va a Mitolario in
italiano
Va a Mitolario italiano -
inglese
Va a Mitolario inglese -
italiano