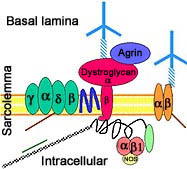
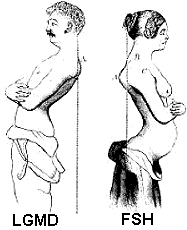
Collegamenti: Leiden
LGMD 1D:

l
Cromosoma 7q; Dominante
-
Nosologia: Confused terminologia
-
Generale: 2 famiglie descritti
-
Clinica
-
Insorgenza: Media 38 anni
-
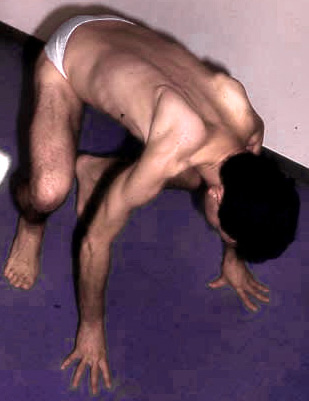
Debolezza: Prossimale > distale; Gambe > Braccia
-
Disfagia (20%)
-
No associata caratteristica sistemiche
-
Laboratorio
-
CK Serico: Normale a 3x elevata
-
Patologia muscolare
-
Miopatica
-
Fibre muscolari sottili rounded e angulated
-
Fibrosi endomisiale
Familiare Cardiomiopatia dilatativa
con conduzione Difetto e Muscolare
Distrofia (?LGMD 1E)
l
Cromosoma 6q23; Dominante
-
Nosologia: Confused terminologia
-
1 grossi famiglia descritti: franco-canadesi discendenti
-
Clinica
-
Debolezza
-
Insorgenza: 2a decade e piů tardi
-
Distribuzione: Prossimale;
Faccia normale
-
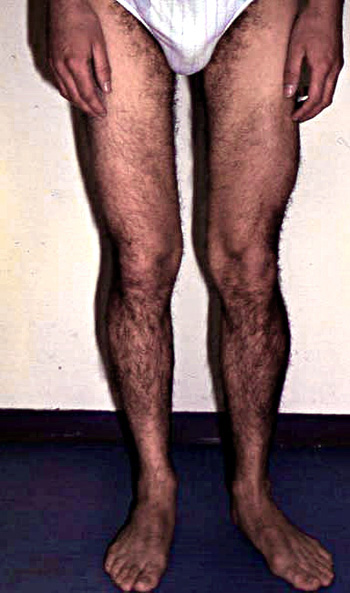
Polpaccio Ipertrofia: Occasionali
-
Progressione: Lenta; Tutti pazienti rimanere ambulatorio
-
Cardiaco
-
Aritmia: Earliest caratteristica del Cardiaca malattia; Insorgenza 2o a 25 anni
-
Insufficienza cardiaca congestiva: 4 camera ingrandimento; Insorgenza 3a to 5a decade
-
Morte improvvisa senza prima sintomi cardiaci
-
Laboratorio
-
Patologia muscolare
-
Fibre di dimensioni variabili
-
Aumentato tessuto connettivo endomisiale
-
CK serico
-
Solitamente aumentati 2x to 4x controllo
-
Piů probabilmente alto nei maschi che femmine
LGMD 1F35
l
Cromosoma 7q32.1-32.2; Dominante
-
1 grossi famiglia descritti: spagnoli
-
Insorgenza
-
Media: 16 anni
-
Gamma: 1 a 58 anni
-
? Anticipazione
-
Prossimale gambe debolezza
-
Debolezza
-
Distribuzione
-
Prossimale: all'inizio del decorso della malattia;
-
Distale: Tarda; Estensore digitorum, Tibiale anteriore, Dito estensori
-
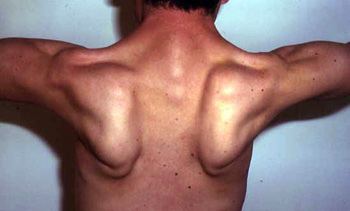
Scapole alate: Alcuni pazienti
-
Gambe > Braccia
-
Faccia: Normale in maggior parte; Alcune con insorgenza piů precoce malattia
-
Respiratorio: Insorgenza piů giovanile pazienti
-
Simmetrica
-
Progressione: Linear rate
-
Piů rapida in giovanier insorgere pazienti
-
Carrozzina nella 2a o 3a decade in giovanier insorgere pazienti
-
Ipertrofia muscolare: Assenti
-
Contratture: Occasionali piů tardi; Caviglie
-
Spinale deModuloity: Alcune giovanier insorgere pazienti
-
Laboratorio
-
Patologia muscolare
-
Fibre di dimensioni variabili
-
Occasionali degenerazione
-
Aumentato connettivo tessuto
-
Desmina: lievemente aumentati espressione in alcuni fibre
-
Vacuoli orlati: Alcuni pazienti
-
Ultrastrutture: Vacuoli autofagici
-
EMG: Miopatica
-
CK serico
-
Normale in 40%
-
Gamma: 47 a 2,920
-
Cardiaco: Normale ECG e Ecocardiogramma
LGMD 1G
l
Cromosoma 4p21; Dominante
-
Epidemiologia: brasiliana-Caucasici famiglia
-
Insorgenza
-
Etŕ: 3o a 47 anni
-
gamba debolezza
-
Crampi alle gambe
-
Clinica
-
omogeneo fenotipo
-
Debolezza
-
Prossimale
-
Gambe 100%; Braccia 90%
-
Atrofia muscolare
-
Contratture: Limitato flessione di dita e pollici
-
Progressione: Lenta; 50% camminare a 10 anni dopo l'insorgenza
-
Laboratorio
-
ECG: Normale
-
Mano x-rays: Normale
-
CK Serico: Solitamente alto (Fino ai 9x normale); Normale in 20%
-
Biopsia muscolare
-
Variazione di dimensione delle fibre
-
Perimisiale fibrosi (Discrete)
-
Necrotica fibre
-
Vacuoli orlati: Colorazione con distrofina e sarcoglycans
-
Sottili atrofica angulated fibre: sparse gruppi
LGMD2: Recessiva Ereditarietŕ
-
Generale
-
Portatori: Have aumentati CK serico in < 5% dei casi
-
geografica foci
LGMD 2A
l
Calpain-3 (p94 proteina)

 ;
Cromosoma 15q15.1-q21.1; Recessiva
;
Cromosoma 15q15.1-q21.1; Recessiva
LGMD 2B
l
Dysferlin
; Cromosoma 2p13.3-p13.1; Recessiva
-
Gene
-
Gene mutaziones25
-
Tipi di mutazioni
-
Localizzazione delle mutazioni in gene: Largamente dispersed over codificante sequenza
-
Variations nel fenotipo non ben spiegate con mutazione tipo
-
geografica distribuzione
-
Le mutazioni presente a basso frequenza in molti popolazioni: Tipicamente
-
1% di recessiva LGMD
-
33% di distale miopatie
-
Comune nella Libyan Jews: 1624delG; Frequenza dei portatori fino a 10%
-
giapponese
-
G3370T: Malattia fenotipo lieve; Insorgenza piů tardiva
-
G3510A: Malattia piů grave; Piů precoce insorgere; CK alto
-
Altro comuni mutazioni: 3746delG; 4870delT
-
Sueca, Spain: Arg1905X50
-
Database delle mutazioni: HGMD
-
Proteina: Dysferlin
-
Clinica: fenotipo lieve
|
LGMD 2B
No polpaccio Ipertrofia
|
-
Insorgenza
-
Etŕ
-
Media 19 e#177; 3 anni
-
Gamma 12 a 39 anni
-
L'eterogeneitŕ in singolo famiglia Puň avvenire
-
gamba debolezza
-
Debolezza:
-
Lieve
-
Gambe
-
Specialmente gastrocnemio
-
Debolezza prossimale: Specialmente quadricipite, anche psoas e quadricipite
-
inferiori arto 9 anni prima superiore arto
-
Braccia: Specialmente bicipite
-
Perdita di ambulation > 30 anni
-
Ampia inter- e intrafamiliare variazione
-
Progressione: Lenta; La maggior parte di camminano fino alla > 33 anni, alcuni dentro 6a decade
-
Polpaccio dimensioni
-
Ipertrofia: Insolita
-
Occasionali distale insorgere pazienti
-
Crampi e Sofferenza muscolare: Alcuni pazienti
-
No cardiomiopatia
-
Stessa mutazioni Puň anche produce Miyoshi distale miopatia
-
? Modifier geni determine fenotipo
-
Caratteristiche di laboratorio
-
CK
-
Tipica: Molto alta; 10x to 72x normale
-
La piů bassa: 343
-
Presymptomatic: 343 a 3.000
-
? Etŕ-dipendente aumento
-
EMG: Miopatica
-
Unitŕ motorie: Piccola ampiezza; Breve durata
-
No spontaneo attivitŕ
-
CT: Gaatrocnemio atrofia
-
Muscoli
|
Dysferlin Colorazione
in normale Muscoli
|
-
Sarcoglicani e distrofina presente
-
Sindromi varianti: differenti fenotipi Puň si trova nella stessa famiglia
-
Miopatia distale
-
Miyoshi
-
Distale anteriori dysferlinopathy
-
Insorgenza: piedi drop; Steppage andatura
-
Debolezza
-
Gambe
-
Anteriori: Caviglie > ginocchia
-
Distale > prossimale
-
Braccia: Prossimale
-
Asintomatica con elevata serico CK livelli
-
Modello di topo: SJL
-
171-bp delezione in Dysferlin gene
-
Dysferlin: Ridotti to 15% del normale
-
Patologia: Miopatia e#177; Infiammazione
LGMD 2C; Gravi Infanzia Autosomica recessiva (SCARMD)
l
γ Sarcoglycan
; Cromosoma 13q12; Recessiva
-
γ Sarcoglycan gene
-
promotores: Muscoli, Cervello e ? Il cuore-tipo
-
Le mutazioni
-
γ Sarcoglycan proteina
-
Caratteristiche cliniche
-
Gravitŕ
-
Alcune con Duchenne-simili decorso
-
Altre intermedio fra Duchenne e Becker fenotipo
-
Lieve Becker fenotipo in occasionali famiglia
-
Variabile: Interfamilial; intrafamiliari puň essere omogeneo o eterogenea
-
Gypsies con C283Y mutazione
-
Precoce Duchenne-simili decorso ma piů lunga sopravvivenza (50%)
-
Becker fenotipo (13%)
-
Generale: Piů grave che North africani con fuori dai frame del521-T mutazione
-
Debolezza
-
Insorgenza
-
Media 5 ai 6 anni
-
C283Y mutazione: < 2 anni
-
Prossimale > distale
-
Irregolare distribuzione con alcuni mutazioni (C283Y)
-
colpiti: Glutei, Adductors, Tendini posteriori delle ginocchia, Spinalis, Abdominals, Subscapularis, Soleo
-
Risparmiati: Quadricipite
-
Respiratorio: Insufficienza in 3a decade
-
Ipertrofia muscolare: Alcuni pazienti (C283Y); Polpaccio e Lingua
-
Perdita di udito: Neurosensory
-
Cardiaco
-
Occasionali coinvolgimento: Specialmente piů tardi in decorso della malattia
-
C283Y mutazione: Subclinical; ECG cambiamenti 60%; RVH; Diastolica disfunzione
-
Scheletro (C283Y): Lombare hyperlordosis; Scapole alate
-
Intelligenza: Normale
-
Decorso
-
Perdita di ambulation: 1o a 37 anni; Media 16 anni
-
No correlazione fra gravitŕ della malattia e etŕ di insorgere
-
Morte: Comune nella 2a decade
-
Caratteristiche di laboratorio
-
CK Serico: Molto alta
-
Patologia muscolare
-
Miopatica
-
Infiammazione: Occasionali
-
Sarcoglycan Colorazione
-
Malattia grave: Assenti γ-sarcoglicani
-
Lentamente progressive malattia: Ridotti γ-sarcoglicani
-
Altro sarcoglycans: Livelli non correlate a cliniche fenotipo
-
α: Normale o ridotta
-
δ: Normale o ridotta
-
β: Variabile; Puň essere ridotta maggior parte o assente
-
Distrofina: Puň essere normale o ridotta
LGMD 2D
l
α-Sarcoglycan (Adhalin)
;
Cromosoma 17q21; Recessiva
LGMD 2E
l
β-Sarcoglycan
;
Cromosoma 4q12; Recessiva
-
Epidemiologia: Comune nella Northern e Southern Indiana Amish; Bern, Svizzera
-
β-Sarcoglycan gene
-
promotores: 1
-
Le mutazioni: Missenso (Comune); Anche nonsenso, inserzioni e delezione
-
database: Leiden;
HGMD
-
β-Sarcoglycan proteina
-
43 kD distrofina-associata glicoproteina
-
Peptide dimensioni: 34.8 kDa
-
Legami alla Distroglicano β by dominio extracellulare
-
Sarcoglycan legante: Fortemente to δ; Moderatamente to γ
-
Localizzazione: Subsarcolemmal
-
Espressione modelli
-
Muscoli: Scheletro e Cardiaco
-
Solo sarcoglicani espresso sul lato esterno Muscoli: Cervello; Reni
-
Vedi: Sarcoglycan complessi
-
Caratteristiche cliniche: Gravi; Occasionali fenotipo lieve
-
Etŕ di insorgenza
-
< 3 anni to Adolescenti
-
Adolescenti insorgere: Da simile a "outlier"
dystrophinopathy
-
Variabilitŕ intrafamiliare
-
Miopatia
-
Debolezza prossimale
-
Ipertrofia muscolare: Preminente; e#177; Lingua
-
Spalle: Scapole alate; Deperimento muscolare
-
Progressione: Spesso in carrozzina by 1o a 15 anni, solitamente by 25 anni
-
Cardiomiopatia
-
Pazienti occasionali
-
Mutazione: 8 bp duplicazione nell'esone 3 di 1 allele
-
Caratteristiche di laboratorio
-
CK serico: Alto; Spesso > 5.000
-
Biopsia muscolare
-
Miopatica
-
Sarcoglicani: Tutti solitamente assente
-
Distrofina: Spesso ridotta ma non assente
-
Cardiaco Muscoli: Sarcoglicani ridotta con la cardiomiopatia
-
β-Sarcoglycan topi knockout
-
Miopatia con Ipertrofia muscolare
-
Cardiomiopatia con focalee aree di necrosi
-
Ridotti Sarcoglycan-sarcospan complessi in muscolatura liscia vascolare:
Vascolare δ in cuore, diaframma e reni
LGMD 2F
l
δ-Sarcoglycan
;
Cromosoma 5q33-34; Recessiva
-
δ-Sarcoglycan gene
-
promotores: 3; 1 in modo predominante nei muscoli scheletrici
-
Le mutazioni
-
Frameshift e#174;
Prematura troncatura della proteina: del656C: africani-brasiliana ancestry
-
Missenso: Glu262Lys (Vicino cisteina-ricchi di regioni)
-
Nonsensi e#174;
Prematura troncatura della proteina: E93Ter; Arg165Ter; Trp30Ter
-
database: HGMD;
Leiden
-
δ-Sarcoglycan proteina
-
Clinica: Fenotipo grave con maggior parte mutazioni producente miopatia
-
Insorgenza: 2 a 10 anni
-
Debolezza: Prossimale; Simmetrica
-
Altri Alterazioni muscolari: Polpaccio Ipertrofia; Crampi
-
Progressione: Carrozzina 9 to 16 anni in maggior parte; Uno paziente camminare a 19 anni
-
Morte: 9 to 19 anni
-
Intelligenza: Normale
-
Cardiaco
-
Clinica variante: Lieve LGMD fenotipo descritti
-
Caratteristiche di laboratorio
-
CK serico: Alto; 10x to 50x normale
-
Biopsia muscolare
-
Miopatica: Degenerazione e rigenerazione delle fibre muscolari
-
ImmunoColorazione
-
δ-Sarcoglycan: Assenti
-
Altro sarcoglycans:
α e β assente;
γ ridotta o assente
-
Distrofina: Presente ma ridotta
-
Modello animale
-
Syrian criceto cardiomiopatia: Delezione in promotore regioni di
δ-Sarcoglycan gene
LGMD 2G
l
Telethonin (Titin-Cap; TCAP)
;
Cromosoma 17q11-12; Recessiva
-
Genetica
-
Le mutazioni puntiModuloi o tight delezioni
-
Le mutazioni produce stop codoni
-
Telethonin (Titin-Cap) proteina
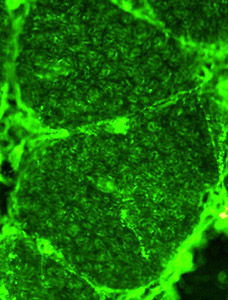
Telethonin nelle fibre muscolari
|
-
funzione
-
Substrato di serina chinasi dominio di titin
-
Titin fosforilates Terminale C dominio di telethonin
-
Succede in iniziale differentiiating miociti
-
Tessuto localizzazione: Cardiaco e muscoli scheletrici
-
Localizzazione cellulare: Sarcomeric banda Z
-
Epidemiologia: Brazil
-
Caratteristiche cliniche
-
Insorgenza: Etŕ media alla 12.5 anni; Gamma 9 to 15 anni
-
Debolezza:
-
Braccia: Prossimale
-
Gambe: Prossimale e distale (piedi drop)
-
Faccia e Collo: Normale
-
Progressione: 40% non-ambulatorio in 3a o 4a decade
-
Dimensioni muscolari
-
Polpaccio Ipertrofia 50%
-
Polpaccio atrofia 50%
-
Coinvolgimento cardiaco 55%
-
Laboratorio
-
CK serico
-
Aumentato 3- to 30-fold
-
inferiori con crescere dell'etŕ
-
Patologia muscolare
-
Miopatica: Degenerazione e rigenerazione
-
Vacuoli orlati
-
Telethonin assente dai muscoli
-
Vedi anche: Cardiomiopatia dilatativa 1N (DCM 1N)
LGMD 2H: Manitoba Hutterite Distrofia
l
Tripartite-motivo contenente gene 32 (TRIM32)
; Cromosoma 9q31-q33; Recessiva
-
Le mutazioni genetiche
-
Omozigote per D487N mutazione puntiforme: Tutti pazienti
-
Mutazione localizzata in NHL dominio
-
StessUna mutazione come Sarcotubulare miopatia
-
Proteina
-
funzione
-
E3-ubiquitina ligasi
-
Labeling delle proteine con ubiqutin per proteosomi degradazione
-
Omo-multimer: Auto-associazione via CC regioni
-
Concentrata in citoplasmatiche e nucleari corpi
-
Tessuto localizzazione: Muscoli; Testicoli; Il cuore; Cute
-
Clinica
-
Variabile fenotipo
-
Etŕ di insorgenza: 8 a 27 anni; Alcune asintomatico in 3a decade
-
Insorgenza: Debolezza prossimale; Schiena dolore; Fatica; Andatura cambiamenti
-
Debolezza
-
Progressione: Lenta; Ambulatory > 50 anni di etŕ
-
Laboratorio
-
CK serico: Alto; 25o a 4,500 U/L
-
EMG: Miopatica
-
Biopsia muscolare: Lieve alterazioni distrofiche
-
Variazione di dimensione delle fibre
-
Degenerazione delle fibre muscolari e rigenerazione
-
Altro: Fibre splitting; Nuclei interni
-
Fibrosi endomisiale
Sarcotubulare miopatia46
l
Tripartite-motivo contenente gene 32 (TRIM32)
; Cromosoma 9q31-q33; Recessiva
-
Epidemiologia
-
Hutterite fratelli dal South Dakota
-
Non-Hutterite fratelli dal tedeschiy
-
Genetica
-
StessUna mutazione come LGMD 2H: Omozigote missenso (D487N)
-
Clinica
-
Insorgenza: Congenita, Infanzia o Adulti (31 anni)
-
Debolezza
-
Prossimale: Simmetrica; Lieve
-
Faccia: Lieve
-
Dimensioni muscolari
-
Ipertrofia: Polpaccio
-
Atrofia: Prossimale
-
Mialgie: Indotta da esercizio
-
Scapole alate
-
Riflessi tendinei: Normale o ridotta
-
Patologia
-
Vacuoli
-
multipla tight rounded
-
In tipo 2 > le fibre di tipo 1
-
reticolo sarcoplasmatico membrane
-
Fibre di dimensioni variate
-
banda Z streaming
LGMD 2I18
l
Fukutina-Proteina relativa gene (FKRP)
; Cromosoma 19q13.3; Recessiva
-
Epidemiologia
-
> 40 familes
-
portatrice rischio: ~ 1:400
-
Comune nella Danimarca e parti di Inghilterra
-
Genetica
-
Le mutazioni puntiModuloi maggior parte comuni: Missenso
-
Mutazione comune in 1 allele (> 90%): Leu276Ileu (C826A)
-
Gravitŕ della malattia correlata con mutazione nella 2a allele
-
Leu276Ileu Omozigotosi correlata con meno fenotipo grave
-
Allelica con
-
FKRP proteina
-
Ubiquitaria
-
Il piů alto livelli: Muscoli scheletrici; Il cuore; Placenta
-
Subcellulare
-
Golgi
-
Muscoli scheletrici e cardiaco
-
Surrounds myonuclei
-
No cambiamenti in locazione con la malattia mutazioni
-
Assenti dalla nuclei di interstiziale cellule
-
Obiettivoed by terminale N e tm dominio
-
Non secreta
-
Glicosiltrasferasi
-
Colocalizzato con α-Mannosidase II
-
Associata con distroglicano processamento
-
Insorgenza
-
Etŕ
-
Debolezza: Camminata ondeggiante; Difficoltŕ con scale; Ipotonia
-
Clinica
-
Debolezza
-
Prossimale > distale
-
Gambe e Braccia
-
Gambe: Cosce adduttori; Psoas; Quadricipite
-
Braccia: Periscapular; Deltoide; bicipiti; tricipite
-
Faccia: Debolezza lieve in alcuni pazienti piů anziani
-
Insufficienza respiratoria (30%)
-
Puň avvenire mentre paziente still ambulante
-
DMD-fenotipo: Insorgenza nella tarda adolescenza
-
Gravitŕ
-
Variabile: DMD-simili a Miopatia ad insorgenza adulta
-
Variabilitŕ intrafamiliare descritti
-
Progressione
-
Insorgenza precoce: Non-ambulante nell'adolescenza
-
Insorgenza piů tardiva: Lenta; Variabile; Alcune (28%) non-ambulante by 4a to 6a decade
-
Dimensioni muscolari
-
Degenerazione: In regioni di debolezza; Braccia > Gambe
-
Ipertrofia: Calves 77%; Cosce;
Lingua
-
Altro occasionali Caratteristiche muscolari
-
Riflessi tendinei: Preservati; ginocchia lievemente ridotta
-
Intelletto: Normale
-
Scheletro
-
Contratture: Spesso alle caviglie; Piů comuni in non-ambulante pazienti
-
Scoliosi
-
Cardiomiopatia
-
Comune: 30% al 50%; Spesso visto on ecocardiografia
-
Tipo: Dilatate
-
Solitamente lieve
-
ventricolare sinistra insufficienza
-
Puň essere isolati caratteristica: Omozigote C826Una mutazione
-
Cute: Puň essere soffici o hyperextensible
-
Gravitŕ variabile
-
Gravi: Insorgenza < 2 anni; Mai run; Lose ambulation nell'adolescenza
-
piů lievi: Insorgenza piů tardiva; Crampi; Ipertrofia muscolare
-
Coerente fenotipo entro famiglie singole
-
Laboratorio
-
CK Serico: Molto alta; 1.000 a 8.000
-
Biopsia muscolare
-
Dimensioni delle fibre muscolari: Variazione
-
Necrotica e in rigenerazione Fibre muscolari
-
Predominanza di fibre muscolari di tipo 1
-
Tessuto connettivo: lievemente aumentati
-
Ridotti Colorazione per
-
distroglicano α: Specialmente in Malattia grave
-
Perdita selettiva di piů alti MW Modulos
-
Meno grave perdita che nella MDC1C
-
Alcuni pazienti hanno normale Colorazione
-
DDx di ridotta distroglicano α
-
α2-laminin (Moderate perdita): Puň essere assente on Occidente blot
-
Normale distrofina, sarcoglycans e Distroglicano β
-
Conduzione nervosa studi: Normale
-
ECG: Normale
-
MRI: Normale CNS o minima atrofia
Miopatia con merosina anormale (Laminina-2)
4
l
Fukutina-Proteina relativa gene (FKRP)
; Cromosoma 19q13.3; Recessiva
-
Genetica: Allelica con LGMD 2I
-
Clinica
-
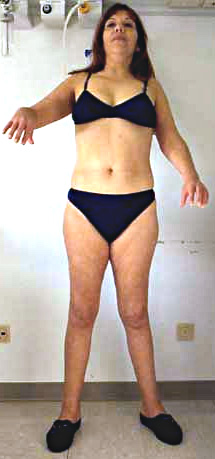
Insorgenza
-
Solitamente adulti: 11 a 40 anni
-
Femmine preponderanza
-
Debolezza
-
Craniale: Occhi chiusura; Collo flessione
-
Estremitŕ superiori: Prossimale; No scapular winging
-
Estremitŕ inferiori: Specialmente hamstrings + anche adductor e flexors
-
Capacitŕ vitale: Ridotti to 30% al 93% del normale
-
Alcune assimetrie
-
Progressiva
-
Ipertrofia muscolare: Calves; Brachioradialis
-
Crampi: Occasionali
-
Scheletro: Lombare lordosi; No contratture
-
CNS (MRI) e cardiaca: Normale
-
Laboratorio
-
CK Serico: 1,100 a 3,700
-
EMG: Miopatica
-
Istologia muscolare
-
Variazione nella dimensione delle fibre
-
Necrosi delle fibre muscolari
-
Muscoli merosina
-
Normale immunocitochimica di muscolo sezione (80 kDa e 300 kDa frammenti)
-
Assenti on Occidente blot
LGMD 2J
l
Titin
;
Cromosoma 2q31; Recessiva
-
Genetica
-
Le mutazioni
-
Omozigote per titin mutazioni
-
Tipi di mutazioni
-
Missenso: Leu293,356Pro (francese)
-
11 bp delezione (Finniche): Final esone (363)
-
Allelica con
-
Clinica
-
Solitamente si ha in famiglie con altre membri aventi dominante debolezza distale sindrome
-
Etŕ di insorgenza
-
Solito: Infanzia
-
Gamma: < 10 anni to 3a decade
-
Debolezza
-
Prossimale > distale
-
Piů grave che nella allelica distale miopatia
-
Faccia: Normale
-
Degenerazione: Anteriori tibial
-
Progressione
-
Solito: Carrozzina < 30 anni, entro 20 anni di insorgere
-
Alcuni pazienti ambulatorio a 60 anni
-
No cardiomiopatia
-
CK serico: Alto
-
Biopsia muscolare
-
Miopatica
-
No vacuoli
-
Perdita del Calpain-3
-
Modello di topo: Omozigote knockout
-
Gravi: Letale
-
Debolezza: Distale > prossimale
LGMD 2K
:
con Ritardo mentale e Ridotti α-dystroglycan45
l
POMT1
;
Cromosoma 9q34.1; Recessiva
-
Epidemiologia: turche famiglie
-
Genetica
-
Insorgenza
-
Etŕ: 1a decade
-
Motorio: Fatica; Difficoltŕ salire le scale ed una correre; Precoce pietra miliare normale
-
Clinica
-
Muscoli
-
Dimensioni: Lieve pseudohypertrophy
-
Debolezza: Prossimale
-
Decorso: Lenta progressione
-
Contrattura delle giunture (50%): Caviglie
-
CNS: Ritardo mentale, IQ 5o a 76
-
Laboratorio
-
CK Serico: Molto alta (9x to 40x elevata)
-
Muscoli
-
Dimensione delle fibre: Variata
-
distroglicano α: Ridotti glicosilazione
-
MRI: Normale CNS
LGMD 2L59
l
Fukutina (FCMD)
;
Cromosoma 9q31; Recessiva
-
Epidemiologia: 2 famiglie, israeliana, e indiane origini
-
Genetica
-
Le mutazioni: 1167dupA; 1363delG; Arg307Gln
-
Allelica con
-
Clinica
-
Insorgenza
-
Etŕ: Meno di 6 mesi
-
Ipotonia
-
Deteriorazione con acute malattia febbrile
-
Debolezza
-
Prossimale e distale
-
Gambe > Braccia
-
Migliora con corticosteroidi trattamento
-
Tutti pazienti ambulante
-
Ipertrofia muscolare: Posteriori gambe e#177; altre
-
CNS: Intelligenza normale
-
Laboratorio
-
CK Serico: Molto alta
-
Biopsia muscolare
-
Morfologia: Distrofica; Necrosi delle fibre muscolari
-
Glicosilata Distroglicano α: Niniziale assente
-
Laminina-α2, β1 e γ1: lievemente ridotta
LGMD 11p1367
l
Cromosoma 11p13-p12; Recessiva
-
Epidemiologia
-
famiglie franco-canadesi
-
Intra-familiare variabilitŕ
-
Clinica
-
Etŕ di insorgenza: Media = 33 anni; Gamma 11 a 50 anni
-
Atrofia muscolare: Quadricipite; bicipiti brachii
-
Asimmetrica
-
Debolezza facciale: Poche pazienti
-
Dolore muscolare: 86%; 50% con, o dopo, esercizio
-
Polpaccio Ipertrofia: Alcuni pazienti
-
Contratture: Insolita
-
Cardiaco: Solitamente normale
-
Laboratorio
-
MRI: Precoce atrofia di mediale quadricipite, hamstrings e adductor magnus
-
CK Serico: Normale a 6000
-
EMG: Miopatica; Alcune con spontaneo attivitŕ
-
Patologia: Muscoli
-
Dimensione delle fibre: Variata
-
Necrosi e rigenerazione
-
Nuclei interni
-
Aumentato tessuto connettivo endomisiale
-
Fibre splitting
-
Infiammazione: In grave caso
Altro Dominante Miopatie
Miopatia di Bethlem
:
Numerose loci
l
Collagene, tipo VI, Subunitŕ α1 (COL6A1)
;
Cromosoma 21q22.3; Dominante
l
Collagene, tipo VI, Subunitŕ α2 (COL6A2)
;
Cromosoma 21q22.3; Dominante
l
Collagene, tipo VI, Subunitŕ
α3 (COL6A3)
;
Cromosoma 2q37; Dominante
-
Le mutazioni genetiche: >10 identificate
-
La maggior parte di mutazioni localizzato fra Esoni 3 e 14
-
La maggior parte di sono aminoacido sostituzioni: Glicina maggior parte comuni (Gli-X-Y)
-
Solitamente in triple elica regioni
-
Alcune mutazioni in globulare regioni
-
Malattia patogenesi: ? Dominante negative effetto
-
Delezione inframe nell'esone 14 di Collagene α1 (IVS14DS, G-A, +1): Mutazione comune
-
Can causano anche malattia by codone di stop
:
Produce aploinsufficienza
-
inferiori incidenza di contratture
-
Altro malattia Correlazione
-
La maggior parte di grave malattia: COL6A1 multi-esone fuori-di-frame delezione; Le mutazioni interno triple elica
-
piů lievi: Esone-skipping mutazioni; Terminale N globulare regioni
-
Allelica con
-
Proteina: Collagene VI
-
Caratteristiche cliniche
-
Insorgenza
-
Etŕ: Variabile entro famiglie
-
Prenatal: Movimenti fetali ridotta
-
Neonatale: Ipotonia
-
Infantile precoce (< 2 anni): Comune con debolezza o Contratture
-
Adulti: Come piů tardi come 6a decade
-
Motorio: Ipotonia
-
Articolazioni: Laxity o Contratture
-
Clinica
-
Debolezza
-
Diffuse; Prossimale > distale
-
Progressiva
-
Solitamente lieve in infantili
-
Puň diventare grave e disabilitante in etŕ adulta
-
Occasionali respiratoria Debolezza muscolare: Casi gravi
-
Falling comuni nei bambini
-
Faccia: Risparmiati
-
Crampi muscolari o dolore: Alcuni pazienti
-
Dimensioni muscolari
-
Degenerazione: Prossimale Muscoli; Distale nelle gambe
-
No Ipertrofia muscolare
-
Contratture
-
Distribuzione
-
Flessione: IP giunture mediale 4 dita, wrists, gomiti, caviglie
-
Prossimale: Pectorals, Ginocchia, Spalle, Anche
-
Troncale
-
Congenita: Torcicollo
-
Malattia progressione: Malattia polmonare restrittiva correlata con debolezza e contratture
-
DorsiFlessione contratture alle caviglie in COL6A2 (G250S) mutazione
-
Decorso
-
Dopo nascita: Spesso spontaneamente migliorano
-
Articolazioni: Alcune sono inizialmente hypermobile Puň sviluppo contratture con il tempo
-
Infanzia e adulti: Puň progresso indipendente di debolezza
-
Alcuni pazienti in famiglie Puň mai hanno contratture
-
No funzionale deterioramento
-
Decorso
-
Precoce: Benigno
-
miglioramento in funzione Attorno pubertŕ
-
Adulti
-
Progressione in debolezza comuni fra 25 e 40 anni
-
Molte carrozzina-legato in etŕ adulta: Solitamente 6a o 7a decade
-
Altre lavoro dentro old etŕ
-
Crampi in alcuni pazienti
-
Cardiaco malattia
-
Diagnosi differenziale:
Emery-Dreifuss MD
-
Laboratorio
-
CK: Normale o elevato to 1,750
-
EMG: Miopatica; Sparse fibrillatons
-
Cardiaco: Normale
-
MRI: T1W sequences31
-
Biopsia muscolare
-
Non-specifico miopatia
-
Variazione di dimensione delle fibre
-
Fibre Splitting
-
Nuclei interni
-
Tessuto connettivo aumentati
-
Lobulated fibre muscolari di tipo I
-
Collagene VI
-
Livelli: Solitamente normale; Puň essere ridotta
-
Localizzazione: Ridotti co-localizzazione cnellamina basale (Collagene IV e Perlecan)
-
Laminina β1
-
Ridotti nelle fibre muscolari lamina basale ma non vasi
-
Puň anche be ridotta nellamina A/C mutazioni
multipla epiphyseal displasia con lieve miopatia13 (EDM3)
l
Collagene, tipo IX, Subunitŕ α-3 (COL9A3)
;
Cromosoma 20q13.3; Dominante
-
Genetica
-
Splice accettore mutazione nell'introne 2
-
Causa skipping dell'esone 3
-
mutante collagene IX č poco cross-collegate
-
Tipo IX collagene proteine
-
Fibril-associata collagene con interrotto triple helices
-
Chain struttura: 4 Noncollagenous domini (NC1-4) separati by 3 triple helical collagenous domini (COL1-3)
-
Localizzazione (Tessuto-specifico isoModulos): Cartilage; Occhi vitreous
-
funzione: Organizzazione e spacing di tipo II collagene fibrille
-
modificazioni: NC3 dominio di 2(IX) catena possono essere modificati da una singolo glycosaminoglycan
-
Clinica
-
Generale
-
Insorgenza: Infanzia; ginocchia dolore
-
? Maschi peggio che femmine
-
Debolezza: Lieve; Simmetrica
-
Collo flessione
-
Braccia: spalle abduzione; Gomito extension
-
Gambe: Prossimale; Tendini posteriori delle ginocchia piů deboli che quadricipite
-
Articolazioni
-
Insorgenza precoce malattia degenerativa
-
Specialmente ginocchia: ginocchia dolore
-
Risparmiati: Anche; Distinguishes malattie dal Ribbing e Fairbank tipi di MED
-
Osteoarthritis
-
Joint spazio narrowing; Femoral condylar bony flattening; Osteophytes e subchondral cisti
-
Insorgenza: Mid-etŕ adulta
-
Scheletro: No bassa statura o brachidattilia
-
Laboratorio
-
CK Serico: lievemente elevata
-
Raggi X: Epiphyseal cambiamenti
-
Biopsia muscolare
-
Lieve miopatici cambiamenti con variabilitŕ nella dimensione delle fibre
-
Perimisiale collagene: Normale
-
ImmunoColorazione: Normale distrofina, merosina e sarcoglycans
-
Epiphyseal chondrocytes: Intracitoplasmatica inclusioni
Dominante Miopatia con Ossa Fragility
51
l
Cromosoma 9p21-p22; Dominante
-
Clinica
-
Debolezza
-
Prossimale
-
Insorgenza: Media 29 anni
-
Progressiva
-
Scheletro
-
Fratture, facile
-
Scarsa healing di lunga ossa
-
Onst: Media 18 anni
-
Altro
-
Cute: Sottile
-
Capelli: Prematura grigia
-
ernias
-
Laboratorio
-
Clotting Malattie
-
EMG: Miopatica
-
Bones: Grossolana trabeculation, Irregolare sclerosi, Corticale Ispessimento, restringimento di medullaria cavities
Dominante Miopatia con
Cardiomiopatia1
l
autosomica dominante
-
Nosologia: ? LGMD 1B
-
Clinica
-
Insorgenza: 4 a 38 anni
-
Debolezza
-
Prossimale; Simmetrica
-
inferiori > estremitŕ superiori
-
bicipiti coinvolti @ 40 anni
-
Contratture: Lieve o assente
-
Cardiaco
-
Disritmia e disturbi della conduzione
-
Morte improvvisa
-
Laboratorio
-
CK Serico: Mediano 3x normale; Gamma - normale to 25x
-
EMG e Biopsia: Miopatica
miopatia dominante con Caviglie contratture e alta CK10
l
Autosomica dominante
-
Clinica
-
Maschi predominanza
-
Insorgenza: 2a o 3a decade
-
Debolezza: Prossimale; Simmetrica
-
Contrattura delle giunture: Caviglie
-
Progressione: Spesso disabili by 6a decade
-
Cardiaco: Normale
-
Laboratorio
-
CK Serico: Molto alta; 12x to 55x normale
-
Biopsia muscolare
-
Variazione nelle Dimensioni delle fibre muscolari
-
Occasionali necrotica Fibre muscolari
Barnes's Miopatia
Altro Recessiva Miopatie
Familiare arti-cingoli Myasthenia
l
Dok-7 (C4ORF25)
; Cromosoma 4; Recessiva
LGMD 4
l
Autosomica recessiva
-
Northern Indiana Amish
-
Collegata to LGMD 2E
Metaboliche miopatie
Infantile autofagici vacuolar miopatia56
l
Autosomica recessiva
-
Epidemiologia: 2 pazienti giapponesi
-
Insorgenza: Congenita Ipotonia
-
Clinica
-
Progressione: Ritardo nello sviluppo
-
Cardiomiopatia: Cardiomegalia
-
Morte: 2 a 30 mesi
-
Muscoli: Vacuoli, autofagici
-
LAMP-2 Colorazione
-
Contiene glicogeno granuli
Infanzia autofagici vacuolar miopatia57
l
Autosomica recessiva o Collegate al cromosoma X portatore
-
Epidemiologia: 1 famiglia; 2 britanniche femmine
-
Insorgenza: 12 anni
-
Clinica
-
Precoce sviluppopment: Normale
-
Debolezza
-
Gambe > Braccia
-
Prossimale > distale
-
Collo flessori
-
Contratture: ginocchia
-
Laboratorio
-
CK serico: Alto, 500 a 700
-
EMG: Miopatica
-
MRI: Degenerazione di coscia Muscoli risparmiando adduttori
-
Muscoli
Insorgenza in etŕ adulta autofagici vacuolar miopatia con multiorgan coinvolgimento58
l
Sporadica
-
Epidemiologia: 1 giapponese maschio
-
Insorgenza: Miopatia come adulti
-
Clinica
-
Oculare
-
Achromatopsia: dal infantili
-
Degenerazione retinica
pigmentaria: Cecitŕ in 3a decade
-
Atrofia ottica
-
Debolezza
-
Prossimale > distale
-
Scapole alate
-
Altri muscoli
-
Exertional dolore
-
Dispnea
-
Cardiomiopatia
-
ECG: Bradicardia con transitoria sinus arresto e prolungato PQ intervallo
-
L'ecocardiografia: Ipertrofia e Parziale dilatazione di ventricolare sinistra wall; Diminuita ejection frazione
-
Polmoni: Fibrosi
-
Laboratorio
-
CK serico: Alto, 500
-
IgG e IgE: alta
-
67Ga apporto nei muscoli scheletrici
-
Muscoli
-
Fegato: Vacuoli in epatociti
-
Renale: Proteinuria
Collegate al cromosoma X miopatie
Collegate al cromosoma X miopatia con eccessiva autophagy5,16
l
Cromosoma Xq28; Recessiva
-
Epidemiologia: 16 famiglie in Europa e nordamerica
-
Genetica
-
Epidemiologia: 14 familes descritti
-
Clinica: Modelli simili fra le famiglie
-
Etŕ di insorgenza
-
Etŕ: Gamma Neonatale to 20 anni; Tipica 5 ai 10 anni
-
Debolezza: Difficoltŕ salire le scale ed una correre
-
Debolezza: arti-cingoli; Lieve
-
Prossimale
-
Simmetrica
-
Gambe > Braccia
-
Distale: Caviglie
-
No Ipertrofia muscolare o cliniche miotonia
-
Progressione
-
Molto lenta o stabile
-
LONGEVITA' non alterato
-
Molte pazienti in carrozzina in 6a decade
-
Cardiaco: Normale
-
CNS: Intelleta normale
-
Femmine portatrici: Normale o lievemente affetti
-
Laboratorio
-
CK Serico: 2x to 15x elevata; Piů alto nell'adolescenza che nellafantili
-
EMG
-
Miopatica
-
Unitŕ motorie: alta ampiezza; Polifasiche; Normale durata
-
Attivitŕ spontanee
-
Miotoniche e alta frequenza scariche
-
Puň trovarsi nella clinicamente non affetti Muscoli
-
La maggior parte di affetti Muscoli: Psoas; Anteriori coscia; Posteriori gambe
-
Biopsia muscolare
-
Fibre muscolari
-
Fibre di dimensioni variabili
-
Necrosi: tight o nessuna
-
Vacuoli: Autofagica con caratteristiche sarcolemmali
-
Eccessiva autofagici attivitŕ
-
Localizzazione: Sarcoplasm; Internal o Vicino superficie
-
Basofile granuli
-
Contenente
-
lisosomiali enzimi
-
Calcio
-
C5b-9 complemento componenti
-
Cellular detriti
-
ImmunoColorazione
-
Distrofina: Vacuolare membrane Colorazione
-
LAMP-2 e Sarcoglycan
-
α1-Antitrypsin: Entro vacuoli
-
Fosfatasi acida: Presenza variabile
-
Vacuolizzate fibre espressa polysialylated isoModulo di NCAM
-
Diagnosi differenziale
-
Exocytosis della phagocytosed materiale
-
lamina basale
-
colpiti fibre circondate by multipla layers
-
Contiene granulare materiale
-
Complementi depositi
-
Sarcolemma e vacuoli
-
granulare Modello
Collegate al cromosoma X Vacuolare Cardiomiopatia e Miopatia (Danon's malattia)
l
Lysosome-associata membrane proteina 2 (LAMP-2)
;
Cromosoma Xq24; Dominante, piů grave nei maschi
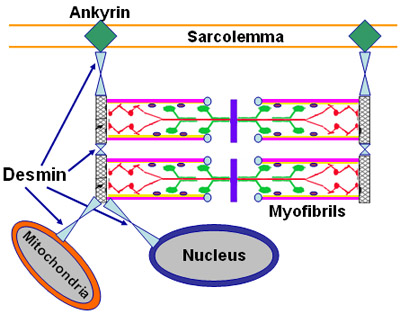
Miofibrillare (Desmina
-accumulo) Myopathies3,11
Miofibrillare miopatie: Generale
-
Caratteristiche cliniche
-
Debolezza
-
Cardiomiopatia (50%)
-
EMG: Miopatia e#177; Denervazione cambiamenti
-
CK Serico: Normale, o elevato < 5x
-
Patologica caratteristiche
-
Degradazione (focalee) primariamente che colpisce miofibrille
-
Lytic lesioni depleto della actina, Actinina α
e#177; titin, nebulina, o miosina
-
Inappropriate espressione di
-
Cell division ciclo (CDC) 2 chinasi
-
Cyclin-dipendente kinases (CDK) 2, 4 e 7
-
ialina spheroid strutture
-
Contiene compacted e degradata miofibrillare strutture
-
React intensamente per Actina
-
Numerosi proteine accumulate in lesioni e Fibre muscolari
-
Desmina, lamin-B, gelsolin, ubiquitina, α1-antichymotrypsin,
NCAM, Terminale N β-AP
-
Ectopic distrofina e γ-sarcoglicani
-
L'espressione ectopica di lamin B e nucleari matrice proteina nel citoplasma
-
amiloide depositi
Miofibrillare miopatie: Sindromi specifiche
Miopatia miofibrillare: Desmina
mutazioni
l
Cromosoma 2q35; Dominante o recessiva
Caratteristiche cliniche
-
Generale
-
Sindromi possono coinvolgere muscoli scheletrici, cuore o entrambi
-
Smooth Muscoli coinvolti in alcuni sindromi
-
eterozigosi: Ala337Pro; Leu345Pro
-
Dominante
-
Meccanismo: ? Dominante negative effetto on filamenti Moduloation
-
Scheletro miopatia
-
Insorgenza: 20's e 30's
-
Precoce: Distale progressione to Debolezza prossimale
-
Progressione di debolezza
-
Arti; Collo; Pettorali; Bulbare e facciale
-
Disabilitŕ: Carrozzina, walker o braces in 75%
-
Insufficienza respiratoria in alcune famiglie
-
Coinvolgimento cardiaco (60%)
-
ECG: a destra blocco cardiaco di branca; ST segmento innalzamento
-
Syncopal episodi: Pacemaker spesso richiede (40%)
-
CK Serico: lievemente elevata nel 50% dei casi
-
EMG: Miopatica; Fibrillazione
-
Patologia muscolare
-
Fibre di dimensioni variabili
-
Nuclei interni: Aumentato
-
Accumulo: Desmina, Vimentin e Nestin accumulate nelle fibre muscolari
-
Localizzazione
-
Citoplasmica
-
Subsarcolemmal e alcuni sarcoplasmici aggregati
-
Accumulazione anche contengono distrofina
-
Aggregati can differenti shapes in tipo 1 e 2 Fibre muscolari
-
-
Ultrastrutture: Granulofilamentous materiale
-
Occasionali: Vacuoli orlati; Sottili bastoncini
-
Composizione eterozigotositŕ (Tipo 3): Ala360Pro e Asn393Ile
-
Recessiva: Heterozygote portatori non coinvolti
-
Cardiaco
-
A-V Blocco di conduzione: richiede pacemaker a all'etŕ di 2 a 10
-
Cardiomiopatia insorgere: Infanzia to 20's
-
Debolezza
-
Prossimale e facciale
-
Insorgenza 20's
-
Respiratorio coinvolgimento
-
Scheletro
-
palato ad arco alto
-
Scoliosi
-
Patologia
-
ialina/desmina plaques (Mallory corpo-simili)
-
Amorphous subsarcolemmali materiale
-
Desmina- e distrofina-immunoreattive
-
Cardiomiopatia
Miopatia miofibrillare: Cristallina e#945;B
(Tipo 1)39
l
Cristallina e#945;B
;
Cromosoma 11q22.3-q23.1
-
Genetica: autosomica dominante
-
Le mutazioni: Miopatia
-
Missenso: R120G
-
Troncamento: Terminale C; 464delCT e Q151X
-
Altre mutazioni nello stesso gene cause: Dominante Congenita Posteriori Polar Catarrata
-
Proteina
|

dal HPRD
Cristallina e#945;B
|
-
Tipo: Colpo di calore proteina 20 (HSP20) famiglia
-
Localizzazione
-
Tissues: Muscoli; Lens; miocardio; Reni epitelium
-
Subcellulare: Cytosol
-
Funzioni
-
Chaperone
-
Lens: Modulos aggregati con αA-crystallin; Mantenimento lens transparency
-
Maggiore componente di fibre Rosenthal nella malattia di Alexander
-
Proteina mutantee
-
Espressa
-
Persino con troncatura mutazioni
-
inferiori livelli del normale proteina
-
Modulos complessies con tipo selvatico proteina
-
Probabilmente esercita effetto dominante negativo
-
Collegamento esterno: HPRD
-
Clinica: Variabile
-
Insorgenza: Precoce to middle etŕ adulta
-
Progressione: Lenta; Improvvisa cardiaca morte
-
Debolezza
-
Prossimale: La maggior parte di pazienti
-
Distale: Gambe; Alcuni pazienti
-
Craniale nervo:
Faccia, Bulbare (velopharyngeal)
-
Respiratorio: 1 paziente
-
Gabbia toracica dolore
-
Cardiaco:
Aritmia, Blocco di conduzione, Insufficienza congestiva
-
Altro sistemi: Lens opacities; intestinale malassorbimento
-
e#177; Neuropatia
-
Laboratorio
-
CK Serico: Normale a 7x normale
-
EMG: Miopatica; Irritable
-
Patologia muscolare
-
Miopatica
-
Fibre di dimensioni variate
-
sparse degeneranti e necrotica fibre
-
Accumulazione di granulofilamentous materiale nei muscoli e cuore
-
Subsarcolemmal e intermiofibrillare
-
Contiene fosforilato desmina
-
molecolare componenti: Desmina, aB-crystallin, Distrofina, NCAM, CDC2 chinasi
-
Molecole non presente: Ubiquitina, Gelsolin, e α1-antichymotrypsin
-
Rubbing fuori dai intermyfibrillar network nelle fibre di tipo I
-
Trichrome: regioni di scuro blue Colorazione
-
Rosso congo: Alcune regioni congophilic
-
Autophagocytosis
-
Nuclei: 7% al 8% mostrare iniziale apoptotico cambiamenti
-
Denervazione: sparse esterasi+ angular Fibre muscolari
-
NOTA: Cristallina e#945;B anche si accumula nelle fibre muscolari in
inclusione corpo miosite
Miopatia miofibrillare con ZASP mutaziones44
l
ZASP
;
Cromosoma 10q22.3-q23.2; Dominante
-
Genetica
-
16 esoni: Numerose tessuto specifico splice varianti
-
Le mutazioni: Missenso
-
A147T e A165V: Miopatia; Vicino o entro motivo Importantee nel collegamento ZASP to banda Z
-
R268C: Esone 9; I piů vecchi una miopatia malattia insorgere
-
Cardiomiopatia: Le mutazioni
-
Spesso in linker regioni fra the PDZ e LIM domini
-
Esone 4: Isolata non-compaction della ventricolare sinistra miocardio (INLVM)
-
Esone 6: INLVM o Cardiomiopatia dilatativa
-
Esone 10: Cardiomiopatia dilatativa
-
Allelica con: Markesbery distale miopatia
-
Locus vicino alla: Miopatia miofibrillare con arrhythmogenic destra ventricolare cardiomiopatia
-
~16% di miofibrillare miopatie
-
ZASP proteina (banda Z alternativamente splicciato PDZ motivo-contenente proteina)
-
Localizzazione subcellulare: Citoplasma
-
Espressione tissutale: Predominantemente nei muscoli scheletrici e cardiaco.
-
Legami alla: Actinina α (Strutturali componente di Z-filamenti che cross-Collegamenti filamenti sottili di adiacente sarcomeri )
-
Struttura
-
Terminale N PDZ dominio: Importante per proteina–proteina interazioni
-
ZASP-simili motivo (ZM motivo): In esoni 4 e 6; 26-residui; Needed per interazione con Actinina α
-
lunga isoModulos: Contiene LIM domini che interagisca con proteina chinasi C sottotipi
-
Splice varianti
-
Muscoli scheletrici: 3 isoModulos
-
piů polmonehe di isoModulo: Lacks esoni 4 e 9
-
2a lunga isoModulo: Lacks esoni 4, 9, e 10
-
Breve isoModulo: Lacks esone 4; Codone di stop nell'esone 9
-
Cardiaco Muscoli isoModulos
-
Spesso contengono esone 4 piuttosto dell'esone 6
-
Le mutazioni nell'esone 6 cause cardiomiopatia negli umani
-
Insorgenza
-
Etŕ: 44 a 73 anni
-
Debolezza: La piů comune iniziale caratteristica
-
Clinica
-
Debolezza: Puň essere prossimale o distale
-
Prossimale > distale (45%)
-
Prossimale + Distale (27%)
-
Distale solo (9%)
-
Prossimale solo (18%)
-
Selettiva scapular: 1 paziente
-
Respiratorio: Non preminente
-
Cardiaco (27%)
-
Polineuropatia (45%)
-
Laboratorio
-
CK Serico: Normale a 6x elevata
-
EMG: Miopatica; Irritable (Fibrillazione e Miotoniche scariche)
-
Patologia muscolare: Miopatia miofibrillare
-
Colorazione tricromica: Pleomorphic hyaline, granular e amorfo depositi
-
Rosso congo: ialina strutture spesso Congofila
-
Anormale fibre regioni wthout ossidativa l'attivitŕ enzimatica
-
Vacuoli: tight, Poche fibre
-
Parziale denervazione e reinnervazione: Alcune biopsie
-
Necrosi o rigenerazione: Comune
-
Cronico cambiamenti: Splitting; Nuclei interni
-
Immunocitochimica: Forte Colorazione per miotilina, desmina, Cristallina e#945;B, distrofina e NCAM
-
Ultrastrutture: banda Z streaming e disintegrazione; Disorganizzata sarcomeri e miofibrille
-
Topo variante: ZASP delezione (Cypher o Oracle)
-
Causa scheletrico e cardiaca miopatia con frammentati bande Z
Miopatia miofibrillare con arrhythmogenic destra ventricolare cardiomiopatia
(ARVD7)14
l
Dominante; Cromosoma 10q22.3
-
Epidemiologia: Svedesi famiglia
-
Simile al loco to: ZASP miopatia
-
Clinica
-
Insorgenza: Adulti; 2o a 60 anni
-
Variabilitŕ intrafamiliare inn gravitŕ
-
Debolezza
-
Lieve: Assiali
-
Moderate: Distale predominante
-
Gravi: Generalizzata
-
Puň essere asimmetrico
-
Cardiaco
-
episodi di gabbia toracica dolore o palpitazioni
-
Cardiomiopatia
-
Aritmia: NonsuColorazioneed tachicardia ventricolare; Atrial flutter
-
ventricolare dilatazione: Specialmente a destra
-
CK Serico: lievemente elevata nel 50% dei casi
-
EMG
-
Miopatica: Prossimale e distale
-
Attivitŕ spontanee: Fibrillazione; Positive picchi d'onda
-
Patologia muscolare
-
Miopatica
-
Dimensione delle fibre: Variata
-
Nuclei interni
-
Tessuto connettivo endomisiale: Aumentato
-
Miofibrillare disorganizzazione (Colorazione NADH)
-
Vacuoli orlati
-
Proteina accumulazioni
-
Desmina accumulo
-
NCAM upregulation: Tutte le fibre in grave pazienti
-
Ultrastrutture
-
Intermyofibrillar accumulazioni di granulofilamentous materiale
-
Miofibrillare disorganizzazione
-
banda Z streaming
-
Occasionali: Corpi citoplasmatici; Vacuoli autofagici
Desmina-associata restrittiva cardiomiopatia28
l
Autosomica dominante
-
Etŕ di insorgenza
-
Variabile; 3 a 92 anni
-
La maggior parte di con insorgere by 42 anni
-
Occasionali portatori asintomatici
-
Cardiomiopatia
-
Restrittiva (Ridotti miocardica compliance)
-
Dispnea
-
Edema: Pleural effusion; Anasarca
-
Venous ipertensione
-
Epatomegalia
-
ECG: Atrial flutter/fibrillazione; ST/Alterazioni nell'onda T
-
Ecocardiogramma: Atrial ingrandimento, sinistra e destra
-
Cardiaco Biopsia: fibrosi interstiziale
-
Muscoli scheletrici: No miopatia
-
Catarrata: Nessuna
-
Desmina accumulazioni: Cardiaco Muscoli; dimostrato con l'immunocitochimica
Cardioneuromiopatia con ialina masses e Bastoncelli nemalinici
l
Autosomica recessiva
-
Insorgenza: Adulti; 5a decade
-
Clinica
-
Debolezza: Distale > prossimale; Insufficienza respiratoria
-
Cardiomiopatia: Sistolica disfunzione; A-V Blocco di conduzione
-
Decorso: Aspettativa di etŕ limitata by respiratoria funzione
-
Laboratorio
-
CK Serico: Normale o alto
-
colonna cervicale: Fusione vertebrale
-
Patologia muscolare
-
Dimensione delle fibre: Variata; Sottili to Ipertrofica
-
ialina masses
-
Localizzazione: Fibre muscolari di tipo I
-
Congofila
-
Contiene: Desmina; αB-Crystallin; Ubiquitina; α1-Antichymotrypsin
-
Bastoncelli nemalinici: Solitamente nelle fibre con hyaline masses
-
Vacuoli
-
Lobulation: Alcune fibre muscolari di tipo I
Desmina miopatia (Tipo 2)
l
Autosomica dominante
-
Insorgenza: Bambino - Adulti
-
Debolezza: Distale e prossimale
-
Faticabilitŕ
-
Disfagia
-
Cardiaco: Occasionali
-
Patologia: Citoplasmica o sferoidi corpi associata con desmina
Distrofia muscolare congenita con inclusioni desminiche
l
Selenoprotein N, 1 (SEPN1)
; Cromosoma 1p36-p35; Recessiva
-
Epidemiologia: Northeastern tedeschiy e polacche famiglie
-
SEPN1 proteina
-
Insorgenza
-
Etŕ: Nascita o Precoce infanzia
-
Ipotonia
-
Clinica
-
Debolezza
-
Prossimale
-
Assiali
-
Faccia
-
Dimensioni muscolari: Sottili
-
Sottile interna cosce
-
diritta calves
-
Piatti piedi
-
Cardiaco
-
a destra ventricolare Ipertrofia
-
Insufficienza cardiaca in 1a 2 decade
-
Normale in alcune famiglie
-
Scoliosi
-
Dorsal lordosi
-
tronco deviation: Laterale
-
Anche: Balanced
-
Rigiditŕ spinale
-
Insorgenza: 8 a 14 anni
-
Contrattura delle giunture: Correlate con il grado della debolezza
-
Decorso
-
Debolezza: Progressiva
-
Morte: Puň trovarsi nella 1a o 2a decade dovuta a insufficienza respiratoria o polmonite
-
Laboratorio
-
CK Serico: Normale o lievemente alto
-
serico aldolasi: Normale o lievemente alto
-
Patologia muscolare
-
Desmina-contenente inclusioni
-
Presente nella 10% delle fibre muscolari
-
Mallory corpo-simili
-
Immunoreattive per: Desmina; Distrofina; Ubiquitina
-
Ultrastrutture: Composed di helical filamenti, 1o a 12 nm diametro
-
Variabilitŕ dimensionale delle fibre
-
Nuclei interni
-
Vacuoli: Occasionali rimmed
-
Tessuto connettivo: lievemente aumentati
-
Predominanza delle fibre muscolari di tipo I
Miofibrillare miopatia47
l
Filamin C (Filamin 2)
; Cromosoma 7q32.1; Dominante
-
Epidemiologia: tedesche famiglia
-
Genetica: Troncamento mutazione
-
Mutazione: W2710X
-
Troncata proteina: Anormale ripiegature; Unable to dimerize appropriatamente
-
Filamin C proteina
-
Clinica
-
Insorgenza
-
Etŕ: 37 a 57 anni
-
Debolezza: Solitamente prossimale
-
Schiena dolore
-
Debolezza
-
Gambe > Braccia
-
Solitamente prossimale > distale
-
1 paziente con selettiva polpaccio debolezza
-
Respiratorio: 50%
-
Polineuropatia: 40%
-
Cardiaco: 1 paziente; Bundle ramo blocco; Ridotti ejection frazione
-
Laboratorio
-
CK Serico: lievemente elevata, 2x to 8x
-
Patologia muscolare: Miopatia miofibrillare
-
Nuclei interni
-
Fibre splitting
-
Necrosi: Isolata fibre
-
Colorazione tricromica di Gomori: ialina masses; Vacuoli
-
Citoplasmica aggregati: Contiene filamin C, desmina, miotilina, distrofina, sarcoglycans
Desmina Malattie: Altro
-
Desmina topi knockout
-
Intolleranza all'esercizio
-
Cardiomiopatia: Dilatate; Cardiomyocyte Ipertrofia
-
Scheletro miopatia
-
Localizzazione: Peso-portante Muscoli e Diaframmatici
-
Anatomia: Perdita di anchorage delle miofibrille to membrana plasmatica a costameres
-
Vasculopatie: inferiori passive e attivi tensione nelle pareti dei vasi
-
Aspettativa di etŕ: Abbreviata
-
rigenerazione: Adipocytes tend to accumulate
-
Desmina-contenente inclusioni
Filamenti intermedi e Malattias20
-
Caratteristiche generali di filamenti intermedi
-
Famiglia
-
~ 65 geni funzionali
-
La maggior parte di sono unica to multicellulari organismi
-
? Ancestor proteina in unicellulari organismo: Crescentin (Nei batteri)
-
Struttura
-
Fibrosi proteine
-
Centrali bastoncelli dominio
-
Coiled coil α-helical
-
Simili struttura in maggior parte IFs: differenti 1° sequenze
-
Heptad ripetizioni: Contiene Acidic residui
-
amino- e carbossilico- termini: vari lunghezze e sequenza
-
10-nm diametro
-
Soluble unitŕ: Tetramero con due antiparallel dimeri
-
Post-traslazionale modificazioni
-
Farnesylation
-
fosforilazione: Regolano
-
Filament organizzazione e solubility
-
Associazione con interActinag proteine
-
Suscettibilitŕ a degradazione durante apoptosi
-
Glicosilazione
-
Transglutamination:
-
Moduloation di protettivi, cornified cellule envelope contribuisce da cute barriera
-
Interazione cons con altre proteine
-
Actina microfilaments (7 nm)
-
Microtubuli (24 nm)
-
Localizzazione
-
La maggior parte di sono citoplasmatiche
-
Lamins nel nucleo
-
Alcune sono proteine integrali di membrana
-
Funzioni: Mediate cellule-tipo-specifico caratteristiche di citoarchitettura
-
Interconnected: Con stessa, o altre acessoria proteine
-
Modulo citoscheletrica scaffolding in cellule eucariote
-
Dynamic: Alcune sono motori proteine
-
No conosciute enzimatici attivitŕ
-
Le mutazioni: Spesso distrutto aggregazione di filamenti intermedi proteine
-
Neurofilamenti
-
Maggiore SE in neuroni: Fino ai 85% della proteina contenuto
-
Composed di 3 subunitŕ
-
NF catena sottile (68 kDa): Modulos spina dorsale e initiates assemblaggio di NF
-
NF medium catena (160kDa)
-
NF spessa catena (200 kDa)
-
Struttura: Carboxy terminali di NFH e NFM Moduloa lato braccia proiezioni
-
Allow interazioni fra le neurofilamenti
-
Underlie the "cross Collegamenti" fra adiacente filamenti
-
Se unassociazione molecole
-
Plectina; BPAG1
-
Mediate the cross-collegando di NF e microtubuli
-
Produce the citoscheletrica lattice
-
fosforilazione
-
Localizzazione
-
4o a 70 Lys-Ser-Pro (KSP) ripetizioni
-
Terminale C domini
-
Probabile ruoli
-
NF assemblaggio
-
NF trasporto dal perikaryon al assoni attraverso le lenta trasporto meccanismi
-
NF incorporazione dentro a citoscheletrica network
-
NF funzione: Determine il calibro assonale e conduzione Velocitŕ
-
Intermediate filamenti proteine e mutazioni
-
α-Internexin
: Neuroni
-
Keratin famiglia: Cellule epiteliali; > 20 proteine
-
Protect dal meccanica e nonmeccanica Modulos di sforzo
-
Numerosi Keratinopathies
-
Epidermolisi bollosa e hyperkeratosis sindromi
-
Corneal distrofia: Keratins 3 e 12
-
Alopecia (Monilethrix): Capelli keratins 1 e 6
-
Pachyonychia congenita: Keratins Keratins 6a, 16, 6b, 17
-
Desmina (2q35): miopatia dominante
-
Desmuslina
:
Muscoli; Co-localizzato con desmina
-
Filensin
: Lens; Catarrata
-
Proteina fibrillare acida della glia (GFAP)
: malattia di Alexander
-
α-Internexin
: CNS
-
Lamins: nucleare
-
Lamina A/C: Emery-Dreifuss distrofia muscolare; differenziate cellule
-
Lamina B
: Tutti cellule tipi
-
Nestin
: Neuroepithelial cellule staminali; cellule muscolari (banda Z); Fibroblasti
-
Neurofilamenti: NF-L; NF-M; NF-H
-
Paranemin
: cellule muscolari; Co-polymer con desmina
-
Peripherins: Neuronali cellule
-
Nervi periferici
: SLA sporadica
-
Photorecettore
: Retinite pigmentosa; Distrofia maculare
-
Phakinin
: Lens; Co-polymer con Filensin
-
Giovanile e Congenita cataratta
-
Syncoilin: Muscoli, Cardiaco e scheletro
-
Synemin
: cellule muscolari; Co-polymer con desmina
-
Vimentin
: Mesenchyme; Null mutazione non ha fenotipo
-
Miofibrillare miopatie: molecolare basi non definiti
-
Intermediate filamenti-Proteine associaate e mutazioni
-
Neurofilament accumulazioni in neuropatia
-
Collegamento esterno: citoscheletro
Citoplasmica BODY MIOPATIE (anche vedi
Miopatia distale)
MIOPATIE CON TUBULAR AGGREGATES
Miopatia con tubulare arrays48
-
Epidemiologia: 3 pazienti: Padre e Son + Sporadica
-
Clinica
-
Insorgenza: 1a to 7a decade
-
Debolezza
-
Gambe > Braccia
-
Prossimale
-
Massa muscolare: Ridotti
-
Sofferenza: Mialgie
-
Laboratorio
-
CK Serico: Normale
-
EMG: Miopatica
-
Patologia muscolare: Inclusioni
-
Eosinofile; lievemente refractile; fibre di tipo 2; Tricromia di Gomori +
-
Ultrastrutture: Arrotondata hexagonal tubulare arrays
Miopatia congenita Sindromi: Insorgenza tardiva o progressive
Modelli animali con Miopatia
-
Tipo XV collagene (COL15A1) Carenza
-
Miopatia: Scheletro e Cardiaco
-
Collapsed capillari: Cellule endoteliali degenerazione
-
Cypher
: Miopatia congenita, grave
-
myd topo
-
Le mutazioni nellaRGE
-
Clinica
-
Anormale andatura e posture, elevata
-
Sorditŕ sensorioneurale
-
Cervello difetti: Da simile a Fukuyama e Muscoli-Occhi encefalo CMD; Tipo II lissencephalic
-
Laboratorio
-
Creatina chinasi serica: alta
-
Patologia muscolare: Distrofica; Ridotti Distroglicano α
Vedi anche: Altro Dystrophies
Ritorno a Indice delle miopatie e giunzioni neuromuscolari
Riferimenti
1. Ann Neurol 1996;39:636-642
2. Current Opinion in Neurologia 2000;13:511-517
3. Current Opinion in Neurologia 1997;10:426-429
4. Cervello 1998;121:581-588
5. Ann Neurol 1988;23:258-65;
Malattie neuromuscolari 2001;11:376-388
6. AJHG 1999;64:788-792
7. AJHG 1999:64:1524-1540
8. Umano molecolare Genetica 1999;8:1329-1336
9. Malattie neuromuscolari 1999;9:308-312
10. Acta Neurol Scand 1999;100:199-201
11. Ann Neurol 1999;46:681-683
12.Neurologia 1999;52:861-863
13. PNAS 2000;97:1212-1217
14. Ann Neurol 1999;46:684-692
15. FASEB J 2000;14:761-768
16. Europ J Hum Genet 2000;8:125-129, Neurologia 2002;59:596–601
17. Umano Mol Genet 2000;9:1453-1459, Hum Mutat 2003;21:473-481
18. Malattie neuromuscolari 2000;10:240-246, Hum Mol Genet 2001;10:2851-2859, Ann Neurol 2003;53:537-542
19. Hum Mol Genet 2000;9:2141-2147;
Cervello 2005;128:2315–2326
20. Curr Opin Cell Biol 2000;12:79-90, N Engl J Med 2004;351:2087-2100
21. Malattie neuromuscolari 2001;11:287-296, pediatrici Neurologia 2001;24:235-237, Cervello 2005;128:732-742
22. Am J Med Genet 2000;91:305-12
23. Neurologia 2001;56:1472-1481
24. Acta Neuropathologica 2001;102:27-35
25. Neurologia 2001;57:271-278, Neurologia 2003;60:1799–1804
26. Nature Genet 2001;28:Settembre
27. Trends Molec Med 2001;7:435-441, Ann Neurol 2003;Online gennaio
28. Clin Genet 2001;59:248-256
29. Neurologia 2002;58:1773–1778
30. Acta Neuropathologica 2002; On-Line Luglio
31. Eur J Paediatr Neurol. 2002;6:309-314, 2002:6:305-307
32. Malattie neuromuscolari 2002;12:984–993
33. GlycoBiologia 2003;13:67R-75R
34. J Clin Pathol 2003;56:624–626R
35. Neurologia 2001;56:450–454,
Neurologia 2003;61:404–406
36. Malattie neuromuscolari 2003; Online Settembre
37. Eur J Paediatr Neurol. 2002;6:309-314
38. Ann Neurol 2003;54:674–678
39. Ann Neurol 2003;54:804-810
40. Neurologia 2004;62:1363–1371
41. Hum Mutat 2004;24:52–62
42. Hum Mol Genet 2004; Online Puň
43. Sperimentale Cell Ricerca 2004;301;1–7
44. Ann Neurol 2005;57:269–276, J Am Coll Cardiol
45. Neuromuscul Disord. 2005;15:271-275
46. Ann Neurol 2005;57:591–595
47. Am J Hum Genet 2005; Online Puň
48. Ann Neurol 1999; 45:512-515
49. Acta Neuropathol (Berl) 2004;107:546-552.
50. Arch Neurol 2005;62:1256-1259
51. Hum Genet. 2005; Oct 22
52. Neurologia 2005;65:1936–1940
53. Neurologia 2006;66:1114-1116
54. Dev Med Bambino Neurol 2006;48:304-306
55. Malattie neuromuscolari 2006;16:S109
56. Neurologia 2001;57;903-905
57. Neuropatologia e Applicato NeuroBiologia 2006;32:253–259
58. Neurologia 2003;61:128–131
59. Ann Neurol 2006; Online Oct 16
60. Malattie neuromuscolari 2006; Online Dicembre
61. JNNP 2006; Online Novembre 6
62. FASEB 2006; Online Dicembre 21
63. Muscoli Nervi 2006;34:656–658
64. Malattie neuromuscolari 2006;16:432–436
65. Circulation 2006;114:2104-2112
66. Umano Mutazione 2005;25:82-89
67. Cervello 2007;130:368–380
26 gennaio 2007
Va a Acronimi e sigle
Va a Mitolario in
italiano
Va a Mitolario italiano -
inglese
Va a Mitolario inglese -
italiano