Traduzione di Natale
Marzari
Traduzione di Natale
Marzari
Home,
Ricerca,
Indice alfabetico,
Collegamenti,
Patologia,
Molecole,
Sindromi,
Muscoli,
Giunzioni neuromuscolari,
Nervi,
Spinale,
Atassia,
Anticorpi e Biopsia,
Informazioni per i
pazienti
|
 |
Traduzione del 16 marzo 2011
ATASSIE EREDITARIE: RECESSIVE, CONGENITE e COLLEGATE A X
Sindromi di atassie recessive
ARCA1 (SCAR8): SYNE1; 6q25
ARCA2 (SCAR9): CABC1; 1q42
Atassia-Aprassia oculomotoria 1 (AOA1): Apratassina; 9p13
Atassia-Aprassia oculomotoria 2 (SCAR1): Senatassina; 9q34
Atassia con neuropatia (SCAN1): TDP1; 14q31
Atassia con paralisi dello sguardo verso l'alto
Atassia simile alla telangectasia (ATLD): MRE11;
11q21
Atassia telangectasia: ATM; 11q22
Cardiomiopatia, dilatativa + atassia:
DNAJC19; 3q26
Atassia Cayman: ATCAY; 19p13
Cerebelloparenchimali disturbi (CPD): II,
III, IV, V
Atassia spastica di Charlevoix-Saguenay: Sacsina; 13q12
Citocromo c ossidasi I: Mitocondriale
Coenzima Q10, carenza: Apratassina; 9p13
Colonna posteriore del midollo spinale + retinite pigmentosa:
1q31
Distrofia maculare
Epilessia + disturbi degli occhi: COL18A1; 21q22
Epilessia e ritardo mentale: 16q21
Atassia di Friedreich: Fratassina (FRDA); 9q13
Atassia di Friedreich 2 (FRDA 2): 9p23
Insorgenza nella fanciullezza (SCAR7): 11p15
Insorgenza infantile, atassia spinocerebellare: Twinkle;
10q24
Insorgenza precoce con riflessi ritenuti (EOCA): 13q12
Intrusioni saccadiche (SCAR4): 1p36
Ipogonadismo
Karak: PLA2G6; 22q13
Lesioni talamiche e atassia ad insorgenza adulta
Leucoencefalopatie con scomparsa materia bianca
Marinesco-Sjögren: SIL1; 5q31
MIRAS:
POLG1; 15q25
Miocloni
Baltico (Unverricht-Lundborg): Cistatina B; 21q22
Epilessia-Atassia: PRICKLE1; 12p11
Movimenti oculari lenti
Paralisi laringea e neuropatia motoria
Perdita uditiva e atrofia ottica (SCAR3): 6p21
Atassia spastica di Portneuf: 2q33
Salla sindrome (Accumulo di acido sialico): SLC17A5; 6q14
SANDO: POLG1; 15q25
SeSAME: KCNJ10; 1q23
Sindrome di Cockayne A ERCC8; 5q12
Sindrome di Cockayne B ERCC6; 10q11
Vitamina E, carenza: Proteina di trasferimento del tocoferolo α ;
8q13
Xeroderma pigmentoso
|
Atassie metaboliche
A-lipoproteinemia β: MTP; 4q22
Acidemia idrossiglutarica L 2
Aciduria 3 Metilglutaconica: DNAJC19;
3q26
Biotidinasi, carenza: 3q25
Carnitina acetiltrasferasi: 9q34
Glutamile γ Cisteina Sintetasi: 6p12
Hartnup: SLC6A19; 5p15
Iperammonemica: Urea ciclo
Ipobetalipoproteinemia: APOB; 2p24, 3p22
Malattia delle urine sciroppose: BCKDH; 19q13
Malattia di Refsum: PHYH; 10pter
Malattia di Wilson: ATP7B; 13q14
Niemann-Pick, Tipo C: NPC1; 18q11
Xantomatosi cerebrotendinea: CYP27; 2q33
Atassie collegate a X
Anemia sideroblastica: ABC7
Arts, Sindrome
CLA2
Congenita
Disturbi extrapiramidali
Ritardo mentale con epilessia: SLC9A6
Pelizaeus-Merzbacher variante allelica: PLP
Piruvato deidrogenasi E1-α
SCAX4
Sindrome di Rett: MECP2
Tremore e declino cognitivo: FMR1; Xq27
Altre sindromi atassiche
Dominante
Collegata a X
Congenita
Riparazione del DNA difetti
Disturbi metabolici mitocondriali
Disturbi multisistemici
Spastica
Acquisite
|
Atassie congenite e disturbi cerebrali
Andatura da dandy: Disturbi cromosomici
ZIC1 e ZIC4; 3q24
FOXC1; 6p25
Aplasia cerebellare e pancreatica: PTF1A; 10p13
Aprosencefalia e disgenesi cerebrale
Atassia cerebellare 1 (SCAR2): 9q34
Atassia cerebellare 3 (SCAR6): 20q11
Atassia-Ritardo mentale: Xq24-q27
Atassia-Sordità: X
Sindrome di Behr
CAMOS (SCAR5): 15q24
COACH sindromi
CC2D2A; 4p15
TMEM67; 8q21
Collegate a X
Atassia + Epilessia: OPHN1; Xq12
Atassia + Ritardo mentale
CASM: Xpter-q13.1
Atassia congenita: Xq23
Hoyeraal-Hreidarsson: DKC1; Xq28
SCAX1: Xp11
SCAX2
SCAX3 (Atassia-Sordità)
SCAX5: Xq25
Displasia cerebellotrigeminaldermica
Distrofie muscolari congenite
Fibroblasti fattore di crescita recettore 3
Gillespie: PAX6; 11p13
Glicoproteine carenti di carboidrati
Hoyeraal-Hreidarsson: Discherina; Xq28
Ipoplasia pontocerebellare
Atrofia cerebrale progressiva (PCH2)
PCH2A: TSEN54; 17q25
PCH2B: TSEN2; 3p25
PCH2C: TSEN34; 19q13
Atrofia muscolare spinale (PCH1): VRK1;
14q32
Infantile fatale (PCH4): TSEN2; 3p25
Insorgenza fetale (PCH5)
Ipoplasia pontocerebellare (PCH6): RARS2; 6
Microcefalia progressiva (CLAM; PCH3): 7q11
Joubert, sindromi
Tipo 1: 9q34
Tipo 2: TMEM216; 11q13
Tipo 3: AHI1; 6q23
Tipo 4: NPHP1; 2q13
Tipo 5: CEP290; 12q21
Tipo 6: TMEM67; 8q21
Tipo 7: RPGRIP1L; 16q12
Tipo 8: ARL13B; 3q11
Tipo 9: CC2D2A; 4p15
Tipo 10: OFD1; Xp22
Lissencefalia
Palatoschisi e ipoplasia cerebellare
Ipoplasia cerebellare: RELN; 7q22
Microcefalia
Malformazione del cervello: CASK; Xp11
Polimicrogiria e agenesi del corpo calloso: EOMES; 3p21
Paralisi cerebrale atassica: 9p12-q12
Pitt-Hopkins: TCF-4; 18q21
Locomozione quadrupede
CAMRQ1: VLDLR; 9p24
CAMRQ2: 17p
CAMRQ3: CA8; 8q11
Ritardo-discinesia-coma: CACNA1A; 19p13;
Dominante |
ATASSIE RECESSIVE
Atassia di Friedreich
 2
2
l
FRDA (Fratassina)
; Cromosoma
9q13-q21.1; Recessiva
l
FRDA2; Cromosoma 9p23-p11; Recessiva
11
- Epidemiologia
- Incidenza: 1 su 30.000 ÷ 50.000
- Frequenza dei portatori: 1 su 60 ÷ 110
- Non trovata nei nativi: Estremo oriente; sub sahariani africani; Australia;
Nativi americani
- 75% delle atassie ereditarie con insorgenza < 25 anni
- Genetica Caratteristiche
- FRDA (Fratassina; X25; FXN) (omologo al lievito
 )
)
- Il gene FRDA contiene 7 esoni: L'esone 6 non è codificante
- Trascrizione: Splicing ® 2 Maggiori sequenze codificanti
- Maggiore: esoni 1, 2, 3, 4, 5a
- Minore: esoni 1, 2, 3, 4, 5b
- Localizzazione
- La più alta in tessuti non nervosi: Cuore; Muscoli scheletrici; Fegato;
Reni; Pancreas
- CNS: Midollo spinale > Cervelletto > Corteccia cerebrale
- La fratassina omologa nel lievito gioca un ruolo nel metabolismo del ferro
- Il ferro si accumula nei mitocondri del lievito mutante
- Possibili meccanismi di malattia
- Il ferro sequestrato non è disponibile per altre ferroproteine
® destabilizzazione
- Attività ridotta delle
subunità contenenti cluster Fe-S
- Subunità localizzate nei complessi mitocondriali I, II, e III e nella aconitasi
- Accumulazione di specie
reattive di O2
® stress ossidativo
- FRDA (Fratassina) malata difettosa, non completamente carente nella
funzione
- Eccesso di ripetizioni della tripletta (GAA) del DNA:
Mutazione solitamente (> 90%)
- Atassia di Friedreich: Espansione nel numero delle ripetizioni GAA nell'introne alla fine dell'esone 1
- Numero di copie GAA
- Normale 6 ÷ 34: Bimodale
- 6 ÷ 12 ripetizioni (83%)
- 14 ÷ 34 ripetizioni (17%)
- Atassia di Friedreich
- Gamma: 67 ÷ 1.700
- Più frequentemente: 800 ÷ 1.000 copie
- Eccesso di ripetizioni o mutazioni puntiformi nel 94% dei pazienti italiani con
il fenotipo FA
- Cambiamenti intergenerazionali: Instabili
- La lunghezza delle ripetizioni può espandersi o contrarsi: Con eguali probabilità
- Le lunghe ripetizioni GAA sono instabili durante la meiosi e la
mitosi
- Trasmissione materna: L'aumento o la diminuzione della lunghezza avvengono con eguale frequenza
- Trasmissione paterna: Diminuisce la dimensione della lunghezza delle ripetizioni
- Media delle variazioni: 150 ripetizioni
- Le ripetizioni GAA espanse sopprimono l'espressione del gene FRDA (Fratassina)
- Bloccano l'allungamento trascrizionale
- Le ripetizioni GAA espanse si legano al DNA auto: "Adesivo"
- Associazione di 2 triplette purina-purina-pirimidina (R-R-Y) a pH neutro
- Forma plasmidi superavvolti negativamente
- Forma una struttura ripiegata di DNA non B
- Le ripetizioni GAA portano a
- Rimodellamento e condensazione della cromatina
- Variegazione dell'effetto posizionale (PEV)
- Modificazione posttraslazionale degli istoni
- Acetilazione, metilazione, fosforilazione, ubiquitinazione
- I cambiamenti possono essere invertiti dagli: Inibitori della deacetilasi degli istoni
- FRDA residuale (Fratassina)
- Alcune FRDA residuali (Fratassina) vengono probabilmente espresse in tutti pazienti
- Quantitativo: Proteine mRNA e FRDA inversamente proporzionali alle dimensioni delle espansioni GAA
- Nessuna mutazione omozigotica nulla identificata: ? La perdita completa della FRDA
(Fratassina) è letale
- Mutazioni puntiformi
- Frequenza: 4% ÷ 6% dei pazienti FA
- Descritte 17 mutazioni puntiformi differenti
- Si osservano nei residui conservati con le proteine omologhe del lievito
- Mutazioni per troncamento: Solitamente nell'esone 1
- Mutazioni missenso: Solitamente negli ultimi 3 esoni codificanti il gene per
la FRDA (Fratassina)
- 3 mutazioni puntiformi comuni: Ile154Phe (Sud Italia); M1I; G130V
- Mutazione missenso: Esempi e caratteristiche
- Esone 3 Stop
- Ile154Phe: Esone 4
- La più comune mutazione puntiforme
- Famiglie del sud Italia
- Gravità della malattia simile alla ripetizione omozigotica GAA
- Introne 3: Distrugge l'accettore del sito di splicing alla fine dell'introne
- G130V: Malattia lieve con progressione lenta; Stessa età di insorgenza
- Mutazioni su altri geni
- Mutazioni puntiformi solitamente eterozigotica con ripetizione GAA nel gene
- Nessuna mutazione puntiforme omozigotica ancora identificata
- Correlazioni genotipo-fenotipo
- Caratteristiche cliniche: Correlate con le lunghezze degli alleli GAA mutanti
- Lunghezza correlata inversamente con l'età dell'insorgenza
- Correlazione con alleli più corti
- Fenotipo più lieve e insorgenza tardiva con ripetizioni < 500 paia di
basi in più brevi
- Molte ripetizioni corte
- Malattia ad insorgenza più precoce
- Più cardiomiopatia
- Riflessi ridotti nelle braccia
- Più rapida progressione della malattia: Età più precoce in carrozzina
- Correlazione con la lunghezza degli alleli più lunghi: Se minore di 800 ripetizioni 18
- Media della lunghezza dell'allele GAA più alta con: Diabete; Cardiomiopatia; Scoliosi
- Altre associazioni con lunghezza delle ripetizioni più lunga: Atrofia ottica; Perdita
uditiva
- Si può avere ereditarietà pseudodominante: Dovuta a
- Numero di ripetizioni GAA intermedio (120-156)
- Atassia spastica
- Insorgenza 38 ÷ 45 anni
- Acadiani
- Mutazioni nella metà carbossilica della FRDA matura (Fratassina) (missenso o troncante):
Fenotipo grave
- Mutazioni missenso in metà aminoterminale della FRDA (Fratassina)
- Malattia atipica e più lieve
- Andatura spastica precoce
- Composizione eterozigotica
- Atrofia ottica più comune
- 2° bassi livelli di FRDA residua
- Portatori eterozigoti: Normali
- Malattia più lieve con l'aplotipo mitocondriale U: Minore cardiomiopatia
- Proteina FRDA (Fratassina)
- Proteina mitocondriale
- Localizzazione: Membrana mitocondriale interna
- Funzioni
- ? Necessaria per il mantenimento del genoma mitocondriale
- Coinvolta nell'omeostasi del ferro
- Chaperone del ferro: Meccanismo dei cluster Fe-S; Metabolismo del eme
- Trasporta il ferro dentro mitocondri
- Assemblaggio e trasporto del ferro-zolfo (Fe-S)
- Funzione respiratoria
- Controllo della produzione delle specie reattive dell'ossigeno
- Proteina simile alla Ferritina: Minore funzione
- Causa distruzione nel lievito
- Le molteplici carenze enzimatiche dipendenti da Fe-S: Aconitasi; complessi I
÷ III
- Contenuto di ferro mitocondriale 10 x più elevato
- Espressione
- Localizzazione: Tutti i tessuti
- Mutazioni
- Espansione GAA: Ridotta espressione della proteina normale;
Meno con ripetizioni più lunghe
- Livelli residuali: Necessari per la sopravvivenza
- Proteine correlate
- Peptidasi intermedia mitocondriale (MIPEP)
: Promuove l'accumulazione del ferro mitocondriale in assenza della fratassina
- Processi della peptidasi mitocondriale (MPP; PMPCB)
: Ritaglia la fratassina
a forma matura
- Altri disturbi del metabolismo del ferro
- Malattia di Huntington
- Aceruloplasminemia
- Neurodegenerazioni con accumulazione di ferro nel cervello
- I
: Pantotenato chinasi
2
- II
: Polipeptide
ferritina leggero
- Caratteristiche neurologiche: Sindrome tipica

- Insorgenza
- Età
- Solitamente: < 20 anni, attorno alla pubertà; Tarda 1a decade o Inizio
della 2a decade
- Gamma: 2 ÷ > 70 anni
- Può variare tra fratelli e sorelle
- Atassia: Instabilità nell'andatura
- Scoliosi
- Allelica con la: Sindrome atassica ad insorgenza tardiva
- Caratteristiche neurologiche
- Cerebellare (100%)
- Atassia: Arti e tronco; Andatura (100%)
- Oculare: Scatti ad onda quadra (Comune); Nistagmo (20%)
- Disartria (95%): Insorgenza entro pochi anni
- Disfagia: Successivamente nel decorso della malattia; Liquidi
- Perdita sensoria (~80%): Specialmente vibrazione e posizioni delle articolazioni
- Riflessi tendinei: Assenti (75%) o ridotti
- Segni nel tratto corticospinale
- Risposta dell'estensore plantare (80%)
- Spasticità: Numero di ripetizioni intermedio
- Motorio
- Debolezza (67% ÷ 88%)
- Estremità inferiori
- Può essere a distribuzione "Piramidale"
- Atrofia muscolare: Muscoli piccoli nelle mani e nei piedi
- Atrofia ottica (30%): Può essere o no scompenso visivo
- Corea: Qualche paziente può avere corea senza atassia
- Perdita uditiva: Neurosensoria (20%)
- Autonomo: freddo, gambe e piedi cianotici più tardi nel decorso; Sfinteri
normali
- Cognitivo: Maggiormente preservato
- Elettrofisiologia
- Potenziali dei nervi sensori: Assenti o ridotti
- Motorio: Normali o lievemente ridotti
- NCV: Normale o lievemente ridotta
- Potenziali somatosensori spinali evocati: Assenti
- VEP: Ampiezza ridotta
- MRI del CNS
- Atrofia del midollo spinale: Cervicale
- Nucleo dentato: Gravemente anormale
- Cervelletto: Lieve atrofia più tardi nel decorso
- Normali: Tronco cerebrale, cervello
- Prognosi
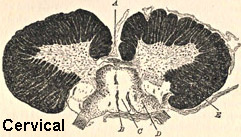
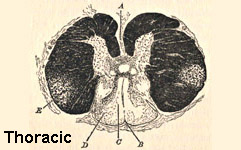
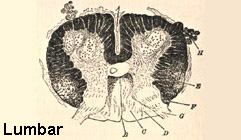
Da Bramwell: Atlante di medicina clinica
Atassia di Friedreich midollo spinale
|
- Decorso
- Progressione lenta
- Tempo alla carrozzina: Media 15 anni; Gamma da pochi anni a decenni
- Femmine: Tempo alla dipendenza dalla carrozzina più breve; Stessa sopravvivenza
- Insorgenza precoce: Tempo alla dipendenza dalla carrozzina più breve
- Morte
- Media: 38 anni
- Gamma: 21 ÷ 69 anni
- Cause: Cardiomiopatia
- Patologia
- Patologia cellulare: Rifluente dagli assoni distali
- Localizzazione
- Midollo spinale
- Tratti spinocerebellari
- Colonna dorsale del midollo spinale
- Tratti piramidali
- Gangli delle radici dorsali: Perdita di neuroni
- Nervi periferici
- Perdita: Assoni sensori, Specialmente grossi mielinizzati
- Atrofia assonale
- Nervi cranici: Coinvolte le
radici entranti
- Cervelletto: Nucleo dentato; Lieve perdita neuronale nella corteccia
- Midollo
- Corteccia cerebrale: Lieve perdita neuronale
- Caratteristiche dei tessuti affetti da FA
- Hanno un quantitativo di fratassina simile a quello di molti tessuti non affetti
- Tipicamente contengono cellule non suddividenti
- Possono dipendere di più dal metabolismo aerobico: CNS; Cuore
- Anormalità biochimiche
- Mitocondri
- Livelli dei fosfolipidi: Ridotti nelle cortecce del cerebellare ed occipitale
- Trattamento: Idebenone per la cardiomiopatia ipertrofica
- Caratteristiche sistemiche
- Scheletrico
- Scoliosi (60% ÷ 80%)
- Piede cavo o equinovaro (50% ÷ 75%)
- Cardiaco (50% ÷ 75%)
- Clinica
- Spesso asintomatica
- Più comune con: Età dell'insorgenza più precoce
- Primi sintomi: Fiato corto; Palpitazioni
- Cardiomiopatia ipertrofica
- ECG: Inversione dell'onda T
- Stenosi subaortica muscolare
- Ventricolo sinistro dilatato ipocinetico
- Onde Q;
Prognosi negativa
- ECG: Anormali (65%)
- Ecocardiografia: Ipertrofia concentrica dei ventricoli; Ipertrofia settale
asimmetrica
- Trattamento
- Idebenone 5-10 mg/kg/giorno all'insorgenza della cardiomiopatia ipertrofica 13
- Gravità: Variabile; Può essere minima o causa di morte
- Endocrino
- Diabete mellito (10%): Specialmente nella FA ad insorgenza < 10 anni
- Intolleranza ai carboidrati (20%): Disfunzione delle cellule β e resistenza periferica all'insulina
- Disturbi degli sfinteri (~25%)
- Anestesia: ? Evitare gli agenti bloccanti la depolarizzazione NM 27
- Atassia di Friedreich: Sindromi neurologiche varianti
- Sindrome atassica a tarda insorgenza
- Insorgenza: Può avvenire tardi fino a 50 ÷ 70 anni
- Alcuni pazienti tarda insorgenza hanno riflessi normali
- Minori deformità scheletriche
- Progressione più lenta
- Atassia spastica: 2 associazioni molecolari
- Numero di ripetizioni GAA intermedio (120÷156): Insorgenza 38
÷ 45 anni;
Acadiani
- Mutazioni missenso in metà aminoterminale della FRDA (Fratassina) in un
allele
- Atassia di Friedreich con riflessi tendinei ritenuti
- Altrimenti tipica sindrome clinica della Fa
- Insorgenza precoce con rapida progressione
- Mutazioni
- Un allele con mutazione missenso nella metà carbossilica della FRDA (Fratassina):
Esone 5a; R165P
- 2o allele tipica espansione GAA
- Sindrome clinica
- Insorgenza precoce: Andatura atassica; 1a decade
- Cerebellare
- Disturbi dell'andatura
- Dismetria: Arti superiori; Lieve
- Disartria: Assente o molto lieve
- Motoneuroni superiori
- Debolezza nelle estremità inferiori
- Dita piegate verso l'alto
- Riflessi tendinei: Braccia normali; Qualche ritenzione degli scatti alle ginocchia
- Coinvolgimento dei nervi periferici: Da leggera a moderata; Potenziali
sensori presenti
- Diabete
- Scheletrico: Piede cavo
- Progressione della malattia: Rapida; Carrozzina nella 2a decade
- Tipo acadiano (forma della Louisiana)
- Epidemiologia: Nordamericani di origine francese
- Mutazioni della fratassina: Numero di ripetizioni intermedio
- Decorso più lieve
- Minore incidenza della cardiomiopatia
- FRDA2
- Epidemiologia: 3 Famiglie consanguinee
- Genetica: Linkage 9p23-p11; Non collegata al gene fratassina
- Caratteristiche cliniche: Simile ala FRDA con mutazioni nel gene fratassina
- Età dell'insorgenza: 5 ÷ 14 anni
- Atassia: Progressiva
- Sensorio: Perdita, specialmente vibrazione e posizione delle articolazioni
- Riflessi tendinei: Assenti o ridotti
- Collegamenti esterni
Atassia con carenza selettiva di vitamina E
l
Proteina di trasferimento del tocoferolo α (ATTP)
; Cromosoma 8q13.1-q13.3
- Mutazioni: Riportate18
- Distribuzione regionale
- Nord Africa: 744delA
- Europa: 513insTT, 486delT e arg134 ÷ ter
- Comuni delezioni con frame shift
- Missenso: Malattia più lieve
- Proteina ATTP: Incorpora il tocoferolo α dentro le lipoproteine secrete dal fegato
- Caratteristiche neurologiche: Fenotipo simile all'Atassia di Friedreich
- Insorgenza
- Fanciullezza: Mutazioni con frame shift
- Mezza età: Mutazione puntiforme (His101Glu)
- nessun segno clinico ma basso livello serico di vitamina E: Eterozigoti
- Perdita sensoria: Modalità a fibre grosse
- Atassia
- Altro sul CNS: Titubazioni del capo 28%; Distonia 13%; Sordità; Disfunzione della vescica
- Riflessi tendinei: Assenti
- Cute: Xantelasmata;Xantomi tendinei
- Retinite pigmentosa: Alcuni pazienti
- Scotoma ad anello
- Elettroretinografia: Nessuna risposta elettrica evocata dalla luce
- Laboratorio
- Siero: Vitamina E moto bassa; Alti colesterolo e trigliceridi
- Elettrodiagnostica: I potenziali sensori possono essere normali
- Trattamento: Integrazione di vitamina E e dieta ricca di grassi
l
A-lipoproteinemia β; Cromosoma 4q24
Atassia spino-cerebellare ad insorgenza infantile (IOSCA)
29
l
Twinkle
; Cromosoma 10q24;
Recessiva
- Epidemiologia: Famiglie finniche
- Genetica
- La maggiore parte delle pazienti omozigoti per la mutazione missenso Tyr508Cys
- Mutazione rara: 1472C->T la regione codificante la transizione silente riduce
la produzione delle proteine Twinkle
- Altre mutazioni causano: Oftalmoplegia esterna progressiva dominante
- Mitocondri
- Normale l'attività enzimatica
- mtDNA: Nessuna delezione o deplezione
- Caratteristiche neurologiche
- Insorgenza: 1 ÷ 2 anni
- Inizialmente
- Atassia
- Atetosi
- Riflessi tendinei: Ridotti
- Successivamente
- CNS
- Oftalmoplegia
- Perdita uditiva
- Attacchi epilettici
- Capacità mentale: Ridotta
- Polineuropatia
- Perdita sensoria: Modalità a fibre grosse
- Motorio: Debolezza distale e atrofia
- Progressiva
- Elettrofisiologia: Ampiezza degli SNAP ridotta
- Patologia nervosa: Perdita di grossi assoni mielinizzati; Sensorio >
motorio
- Prognosi
- Carrozzina nell'adolescenza
- Morte precoce: Correlata al disturbo epilettico
- Sistemico
- Femmine ipogonadismo primario: Ipogonadotrofico
- Nessuna patologia cardiaca
- Patologia del CNS
- Atrofia del cervelletto, del tronco cerebrale e del midollo spinale
- Deplezione del mtDNA nel cervello e nel fegato
- Carenza dei complessi I e IV della catena respiratoria
Baltico Mioclone (Unverricht-Lundborg)
l
Cistatina B
; Cromosoma 21q22.3
- Genetica Caratteristiche
- La mutazione più comune:
Ripetizioni extra di 12 paia di basi ripetute in tandem
- Sequenza ripetuta: (CCCCGCCCCGCG)n
- Localizzazione della ripetizione: regione 5' non traslata
- Numero normale: 2 o 3 ripetizioni
- Pazienti: Inserzione 600 ÷ 900 bp
- Nessuna prova di instabilità intergenerazionale
- Occasionali mutazioni puntiformi missenso
- Nessuna mutazione nulla
- Caratteristiche neurologiche
- Insorgenza 10 ÷ 20 anni
- Epilessia mioclonica
- Atassia: Successivamente nel decorso della malattia
- Morte 25 ÷ 35 anni
- Decorso più lento che nella malattia di Lafora
- Trattamento: Acido valproico; Clonazepam; Fenobarbital
Sindrome epilessia-atassia e mioclone progressivo
46
l
PRICKLE1
; Cromosoma 12p11–q13;
Recessiva
- Epidemiologia: Famiglie del medio oriente
- Genetica: Omozigosi, mutazione missenso; R104Q
- proteina PRICKLE1
- Espressione: Molteplici regioni del cervello
- Neuronale
- Membrana nucleare esterna
- Processi di segnalazione (WNT/PCP) di polarità cellulare non canonica o
planare
- Proteina mutata: Ridotto il legame a REST
- Clinica
- Insorgenza
- Età: da 4 a 5 anni
- Atassia
- Atassia: Disturbi dell'andatura
- Mioclone
- Attacchi epilettici: Insorgenza 5 ÷ 10 anni
- Insufficienza cognitiva: Lievi o nessuna
- Occhi: Ridotto lo sguardo verso l'alto
- Neuropatia sensoria: Alcuni pazienti
- Progressiva
- Laboratorio
- Imaging del cervello: Normali
Marinesco-Sjögren
l
SIL1
Cromosoma 5q31; Recessiva
- Genetica 30
- Tipi di mutazione: Perdita di funzione
- Pazienti finlandesi: Duplicazione omozigotica di 4 nucleotidi; 506_509dupAAGA
nell'esone 6
- Svedesi: Composizione eterozigotica; 506_509dupAAGA e mutazione del sito
donatore di splicing nell'introne 6 (645+2TtoC)
- Altre mutazioni
- Spesso omozigosi; 331CtoT e 212dupA
- Altre: Codoni di stop; Frame shift; Sito di splicing
- Tutti i pazienti con mutazioni hanno miopatia
- Proteina SIL1
- Fattore di scambio nucleotidico per la proteina da shock termico 70 (HSP70)
chaperone HSPA5
- La più alta espressione in organi secretori
- Presente nei muscoli
- Crescita subsarcolemmale spesso vicino ai nuclei
- Localizzazione: Reticolo endoplasmatico rugoso
- Insorgenza: Infanzia
- CNS
- Disfunzione cerebellare
- Disartria; Nistagmo; Atassia
- Atrofia corticale cerebellare: Cellule di Purkinje vacuolate o
binucleate
- Ritardo nello sviluppo: Ritardata maturazione motoria e mentale
- Muscoli
- Sindromi cliniche
- Rabdomiolisi: Dopo infezioni virali
- Debolezza: Progressiva; Alcuni con insorgenza infantile
- Ipotonia
- Creatina chinasi serica (CK): Alta; Elevazione sostenuta o episodica
- Patologia
- Alterazioni miopatiche
- Variazione nella dimensione delle fibre
- Degenerazione e rigenerazione
- Nuclei interni
- Tessuto connettivo endomisiale: Aumentato
- Vacuoli bordati
- Ultrastrutture muscolari
- Vacuoli autofagici con corpi mieloidi
- Nuclei: Patologia più preminente nei muscoli con vacuoli bordati
- Dense strutture membranose nucleari
- Granuli di cromatina condensata
- Vacuolazioni con inclusioni amorfe
- Apoptosi: In mionuclei sparsi
- Proteina SIL1 nei muscoli dei pazienti
- Omozigosi nei pazienti finlandesi
- Immunocolorazione non definita: Compatibile con a prevista perdita di
epitopi
- Composizione eterozigotica
- Colorazione simile ai controlli
- Denervazione: Atrofia a gruppi
- Polineuropatia
- Caratteristiche sistemiche
- Cataratte: Prima fanciullezza o congenite
- Scheletrico: Bassa statura; Cifoscoliosi; Contratture
- Metatarsali e metacarpali corte
- Sterno sporgente
- Ipogonadismo
- ? Alcuni pazienti con malattia di ritenzione chilomicronica e
carenza di vitamina E
- Progressione: Lenta
- Variante: CCFDN
- Sindrome simile con locus sul 18qter: Mutazioni CTDP1
- Differenze dalla sindrome CCFDN
- Presenti nella CCFDN: Neuropatia periferica; Dismorfismo facciale;
Microcornea; Linkage al 18qter
- Presente in MS: Gravità del ritardo mentale; Atrofia cerebellare; Miopatia cronica
- Topo mutante: woozy
Atassia spastica di Charlevoix-Saguenay
l
Sacsina
; Cromosoma 13q12;
Recessiva
- Genetica
- Proteina sacsina
- Epidemiologia
- Localizzazione delle famiglie: Franco-canadesi (Quebec); Tunisia 3
- Decorso clinico
- Insorgenza: Precoce
- Età: Gamma 1 ÷ 20 anni; Media 4 anni
- Più grave e insorgenza più precoce nelle famiglie franco-canadesi
- Nelle famiglie franco-canadesi non camminano mai normalmente
- Atassia: Andatura
- Caratteristiche neurologiche
- Cerebellare
- Atassia: Disartria; Disturbi dell'andatura
- Oculare: Nistagmo (100%); Bassa motilità oculare
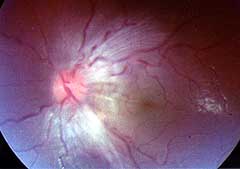
Di H Topaloglu
Fibre nervose mielinizzate nella retina
|
- Occhi
- Coniugati difetti dello sguardo
- Retina: Fibre nervose mielinizzate prominenti (Frequenza variabile)
- Motorio
- Spasticità
- Paraparesi con disturbo dell'andatura: La maggiore parte delle pazienti
- Risposta dell'estensore plantare
- Progressiva
- Nessuna spasticità: Mutazione Phe304Ser omozigotica
- Atrofia distale e debolezza
- Gambe > braccia
- Più apparente nei pazienti più anziani
- Sensorio: Perdita delle sensazioni profonde
- Riflessi tendinei: Vivaci eccetto alle caviglie
- Vescica: Stimolo nel 65%
- Sistemico
- Prolasso della valvola mitrale
- Piede cavo e dita dei piedi a martello: Alcuni pazienti
- Progressione
- Molto lenta; Piccole alterazioni dopo 20 anni
- Variabili fra membri della famiglia
- Alcuni pazienti in carrozzina a 30 anni
- Laboratorio
- Elettrofisiologia
- NCV
- Potenziali di conduzione dei nervi sensori: Assenti
- CMAP: Piccoli
- Velocità di conduzione: Leggermente lenta
- EMG: Denervazione dei muscoli distali
- Potenziali sensori evocati: Anormali
- Biopsia dei nervi
- Perdita di grossi assoni mielinizzati
- Gruppi rigeneranti
- Occasionali bulbi a cipolla e assoni sottili mielinizzati
- Muscoli: Atrofia neurogena
- Risposte visive evocate: Ritardate
- Scansione CT: Atrofia cerebellare del verme superiore e dei lobi anteriori
- Patologia del CNS
- Atrofia cerebellare del verme superiore e dei lobi anteriori
- Assenza delle cellule di Purkinje
- Sindromi varianti
- Atassia senza spasticità o assoni retinici mielinizzati: Mutazioni nonsenso
omozigotiche, Arg2119X
- Atassia senza spasticità
Atassia Cayman
l
ATCAY (Caytaxina)
; Cromosoma 19p13.3;
Recessiva
- Mutazioni
- Ser301Arg
- G-T sostituzione in 3a base dell'introne 9
- Proteina caytaxina
- Espresso solamente nei tessuti nervosi: Cervello; Gangli delle radici dorsali; Neuroni enterici
- Contiene il motivo CRAL-TRIO: Comunemente lega piccole molecole lipofiliche
- Altre proteina con il motivo CRAL-TRIO: ATTP; Causa atassia rispondente alla vitamina E
- Epidemiologia: Comune in 1 regione dell'isola Cayman Grande
- Insorgenza: Ipotonia della prima fanciullezza
- Clinica
- Segni cerebellari: Non progressivi
- Tremore intenzionale
- Parlata disarticolata
- Andatura a gambe larghe
- Nistagmo
- Ritardo psicomotorio: Marcato
- Imaging del cervello: Ipoplasia cerebellare
- Modello di topo: Jittery
Insorgenza precoce Cerebrale atassia con riflessi tendinei ritenuti (EOCA)
l
Cromosoma 13q11-12 + Altre loci; Recessiva
- Genetica
- 13q11-12 locus identificate in 1 famiglia tunisina 3
- Probabile eterogeneità
- Insorgenza: Prima o seconda decade
- Clinica
- Atassia
- Polineuropatia
- Riflessi tendinei: Preservati
- Abilità cognitive e visivo-spaziali: Progressivamente insufficienti
- Progressione
- Minore disabilità che con l'atassia di Friedreich
- Costretti in carrozzina ~ 5 anni dopo l'insorgenza
- Imaging
- MRI: Atrofia cerebellare
- PET: Ridotto il legame al recettore delle benzodiazepine cerebellari
- Elettrodiagnostica: Neuropatia assonale
- Diagnosi differenziale
Atassia cerebellare, ad insorgenza nella fanciullezza (SCAR7)
l
Cromosoma 11p15; Recessiva
- Epidemiologia: Famiglia olandese
- Insorgenza: Prima fanciullezza
- Clinica
- Atassia cerebellare
- Disartria
- Arti: Dismetria; Scrittura anormale
- Disturbi dell'andatura: Alla fine gravi
- Occhi: Nistagmo; Movimenti saccadici
- Piramidale
- Gambe: Spastiche
- Riflessi tendinei: Aumentati nelle gambe (80%)
- Risposta plantare: Estensore (40%)
- Fascicolazioni
- Perdita sensoria (60%): Vibrazioni nelle gambe
- Tremore: Mani, posturale (40%)
- Insufficienza cognitiva: 1 paziente
- Progressione: Lenta
- MRI
- Atrofia del cervelletto e del ponte
Atassia con aprassia oculomotoria 1 (AOA1)9
l
Apratassina
; Cromosoma 9p13;
Recessiva
- Mutazioni del gene
- Proteina
- Membro della superfamiglia della triade istidina
- Espressione: Ubiquitaria
- Localizzazione subcellulare
- Nucleare
- Presente nel nucleoplasma e nel nucleolo
- Ruolo nella riparazione del DNA
- Interagisce con le proteine riparatrici XRCC1, PARP-1 e p53
- Co-localizza con XRCC1 lungo particelle cariche tracciate sulla cromatina
- Influenza la risposta cellulare allo stress genotossico
- Espressa normalmente come forme lunga e corta
- La forma lunga più affetta da mutazione
- La proteina mutata è instabile
- Epidemiologia
- Portoghesi e giapponesi
- La più frequente cause di atassia autosomica recessiva
in Giappone
- Insorgenza: Media 4,7 anni; Gamma 1 ÷ 16 anni
- Clinica
- Atassia (100%)
- Disturbi dell'andatura
- Disartria
- Dismetria degli arti: Braccia > gambe; Più tardi nel decorso
- Extrapiramidale (59%): Distonia (Braccia); Maschera facciale; Occasionalmente
grave
- Oculare
- Aprassia oculomotoria (100%); Insorgenza dopo l'andatura atassica
- Atrofia ottica (14%)
- Sistema nervoso periferico (100%)
- Neuropatia motoria
- Debolezza: Spesso progressiva fino a grave disabilità
- Atrofia
- Distribuzione: Distale; Simmetrica
- Perdita sensoria
- Tarda nel decorso
- Lieve perdita dei sensi vibratorio e della posizione delle giunture
- Altro CNS: Alcuni pazienti, specialmente giapponesi, con ritardo mentale
- Progressione: Lenta
- Laboratorio
- Elettrofisiologia: Neuropatia assonale
- Patologia nervosa: Lieve perdita di grossi assoni mielinizzati
- MRI: Cerebellare ± atrofia del tronco cerebrale
- Ipoalbuminemia
- Livelli del coenzima Q10: Possono essere
ridotti nel muscolo
- Spesso sotto il normale: Nessuna relazione la durata della malattia e la gravità
- Livelli specialmente bassi con la mutazione Trp279Ter omozigotica
- Difetto secondario
- Nessun effetto sui livelli degli enzimi ossidativi mitocondriali
- Vedi: Atassia con carenza di coenzima Q10
Atassia cerebellare con carenza di coenzima Q10
muscolare 1
l
Apratassina (APTX)
; Cromosoma 9p13;
Recessiva
- Epidemiologia: Singola famiglia
- Genetica
- Mutazione: Omozigosi Trp279Ter
- Allelica con: AOA1
- Insorgenza
- Sindromi correlate all'età
- Ipotonia e ritardo motorio: Prima dei 2 anni
- Atassia: Si manifesta tra 2 ÷ 9 anni
- Attacchi epilettici: Età 2 ÷ 6 anni
- Clinica
- Atassia cerebellare (100%)
- Insorgenza in tutti a 10 anni
- Distribuzione: Tronco; Arti; Linguaggio
- Attacchi epilettici (37%)
- Ritardo mentale (30%)
- Segni piramidali (40%): Variabili
- Riflessi tendinei: Vivaci
- Dita dei piedi: Aumentanti
- Paraparesi spastica: Alcuni pazienti
- Debolezza (50%)
- Distale o prossimale
- Anche: Ritardo motorio, sviluppo
- Rabdomiolisi: Alcuni pazienti
- Neuropatia (20%)
- Perdita sensoria
- Assonale
- Altre occasionale CNS
- Emiparesi: Ricorrente
- Oftalmoplegia
- Mioclone
- Sistemico
- Scoliosi
- Ipogonadismo ipergonadotropo: Casi a tarda insorgenza
- Decorso: Progressione lenta ; Carrozzina a 8 anni in alcuni
- Laboratorio
- Muscoli
- Morfologia: Anormalità non specifiche
- Alterazioni di denervazione
- Livelli del Coenzima Q10: 26% ÷ 35% del normale;
Meno di 15 μmol/g tessuto
- Biochimica: Solo il 25% dei pazienti hanno il blocco aspettato nel
trasferimento degli elettroni al complesso III
- Esame sistemico
- Acidosi lattica: Inconsistente
- Normali: Profili degli aminoacidi e degli acidi organici
- Sindrome adulti: FSH e LH alte
- Elettrofisiologia
- NCV: Leggermente rallentata; CMAP piccoli
- SEP: Piccola ampiezza
- Patologia del CNS: Atrofia cerebellare, emisferi e verme
- Trattamento
- Integrazione dietetica con coenzima Q10: 200 mg
÷ 1,200 mg per giorno
- Risposta al trattamento più probabilmente nei bambini che negli adulti
- Vedi anche
SCAR9: Atassia cerebellare, attacchi epilettici e carenza di ubichinone (ARCA2)39
l
Attività di chaperone di BC1 simile al complesso (CABC1, COQ8, ADCK3)
; Cromosoma 1q42.2;
Recessiva o sporadica
- Epidemiologia: Famiglie algerine, francesi e US
- Genetica: Mutazioni
- Tipo: Missenso, sito di splicing, frame shift, delezione
- Localizzazioni: R213W, G272V, G272D, Tyr514Cys, Gly549Ser, E551K,
Gln167X, Thr584del, c.[1812_1813insG], 1398+2T-C,
Asp420X, Lys314_Gln360 del
- Proteina CABC1
- Localizzazione subcellulare: Mitocondri
- Necessario per la biosintesi del CoQ
- Proteina chinasi: Atipica
- Espressione: Ubiquitaria
- Obiettivo del gene soppressore del tumore p53
- Proteina ADCK (Chinasi contenente il dominio aarF)
- Condivide il dominio del terminale N comune: Lys276, Glu278 e Gln279
- Correlata metabolismo dell'ubichinone
- Clinica
- Insorgenza
- Età 1,5 ÷ 11 anni
- Andatura atassica
- Intolleranza all'esercizio
- CNS
- Atassia
- Ritardo mentale: 50%; Da lieve a moderato
- Attacchi epilettici: Alcuni pazienti; Epilessia parziale continua
- Intolleranza all'esercizio (60%)
- Riflessi tendinei: Da ridotti ad aumentati
- Decorso: Progressione lenta (Decenni)
- Trattamento: L'integrazione con coenzima Q10 ha poco o nessun beneficio
- Laboratorio
- Acido lattico: Alto o normale nel siero
- MRI
- Atrofia cerebellare
- Iperintensità su T2 simile a ictus
- EMG: Solitamente normale
- Biopsia muscolare
- Non specifica
- Gocciole lipidiche
- Nessuna fibra rossa sfilacciata
- Enzimi ossidativi mitocondriali: Suggeriscono carenza di chinone
- Alti rapporti di attività CIV/CII+III e CII+III/CII
- Attività degli enzimi nei mitocondri muscolari aumentata dal decilubichinone
- Coenzima Q10:
Basso
Atassia con aprassia oculomotoria 2 (AOA2; SCAR1; SCAN2)
8
l
Senatassina (SETX)
; Cromosoma 9q34;
Recessiva
- Nosologia: Chiamata anche atassia spinocerebellare, recessiva, non di Friedreich,
di tipo 1 (SCAR1)
- Epidemiologia: Comune nei francocanadesi
- Genetica
- Mutazioni
- Terminazione prematura nei 2/3 dei casi
- Missenso in alcune famiglie
- Spesso omozigotica
- Mutazioni missenso in
- Locus vicino alla atassia congenita di Joubert
- Proteina senatassina
- Coinvolta nella maturazione e terminazione del RNA
- Elicasi: Superfamiglia 1
- Espressione: Generalmente ubiquitaria
- Clinica
- Insorgenza: 2 ÷ 22 anni; Media 15 anni
- Atassia
- Disturbi dell'andatura: Gravi
- Arti e tronco: Lievi
- Extrapiramidale
- Coreoatetosi
- Postura distonica nel camminare
- Maschera facciale: Con malattia grave
- Oftalmologico
- Aprassia oculomotoria: Presente variabilmente (47%)
- Scorrimento dello sguardo: Disordinato
- Nistagmo optocinetico: Assente
- Paralisi saccadica
- Saccadi lente: Prima gira in capo, poi seguono gli occhi
- Distribuzione: Globale; Verticale > orizzontale
- Anatomia delle saccadici verticali: Originate vicino ai nuclei interstiziali rostrali
- Neuropatia periferica (93%)
- Perdita sensoria: Vibrazioni (100%); Posizione (74%); Tocco leggero (57%)
- Motorio: Insorgenza 3a decade
- Debolezza: Distale
- Atrofia: Distale
- Elettrodiagnostica: Perdita assonale
- SNAP: Assenti o di ampiezza ridotta
- Motorio: Ampiezza dei CMAP lievemente ridotta
- EMG: Denervazione distale
- Riflessi
- Tendinei: Assenti nelle gambe
- Plantare: Estensore
- Corticale: Intelligenza normale
- Neoplasia: Nessuna associazione
- Progressione
- Nei primi 20 anni, poi stabile
- Disabilità al lavoro
- Diagnosi differenziale: Atassia + insufficienza saccadica
- Laboratorio
- CT: Atrofia cerebellare in alcuni pazienti, lieve
- Conduzione nervosa: Assenti i potenziali sensori
- Fetoproteina a: Alta
- CK serica: Alta in alcuni pazienti
atassia spinocerebellare con cecità e sordità (SCAR3)
l
Cromosoma 6p21; Recessiva
- Insorgenza: Fanciullezza
- Clinica
- Atassia cerebellare
- Atrofia ottica: Perdita visiva
- Degenerazione cocleare: Sordità
Disturbo cerebelloparenchimale II (CPD II)
- Insorgenza: 4a e 5a decade
- Atassia cerebellare pura
Disturbo cerebelloparenchimale V (CPD V; Dissinergia cerebellare mioclonica)
- Insorgenza: 4a e 5a decade
- Atassia; Mioclone
- Patologia
- Perdita del neurone dentato
- Perdita delle fibre del peduncolo cerebellare superiore
Leucoencefalopatie con scomparsa materia bianca
l
Traslocazione iniziazione fattore eIF2B1, subunità α
; Cromosoma 12;
Recessiva
l
Traslocazione iniziazione fattore eIF2B2, subunità β
; Cromosoma 14q24;
Recessiva
l
Traslocazione iniziazione fattore eIF2B3, subunità γ
; Cromosoma 1p34.1;
Recessiva
l
Traslocazione iniziazione fattore eIF2B4, subunità δ
; Cromosoma 2p23.3;
Recessiva
l
Traslocazione iniziazione fattore eIF2B5, subunità e
; Cromosoma 3q27;
Recessiva
l
proteina mitocondriale indotta da MYC
(B17.2L; Mimitina)
; Cromosoma 5q12.1;
Recessiva
l
Dominante
- Nosologia: Altri nomi
- Atassia infantile con ipomielinizzazione diffusa
- Mielinosclerosi centrale diffusa
- Genetica
- ? Allelica con la leucodistrofia infantile fatale
- Identificate ~84 mutazioni
- Gene più comunemente mutato: eIF2B5, R113H
- Fenotipo più leggero (insorgenza > età 5): Mutazioni negli aminoacidi non conservate
(R113H in EIF2B5 e E213G in EIF2B2)
- Fenotipo grave: Mutazioni negli aminoacidi altamente conservate (R195H in
EIF2B5)
- Funzione della proteina eIF2B
- Iniziazione e regolazione della traslazione del RNA dentro la proteina
- Coinvolta specialmente con stress o febbre
- Catalizza lo scambio del 2° legame del fattore di iniziazione
eucariotico GDP per
GTP
- Insorgenza: Variabile
- Solitamente: Fanciullezza
- Occasionali: Insorgenza adulta
- Decorso
- Sviluppo motorio e mentale infantile iniziale: Normale o lievemente
ritardato
- Progressivo a cronico
- Episodica
- Deteriorazione dopo infezioni e traumi minori al capo
- Periodi di coma
- Morte: Dopo anni ÷ decenni
- Segni neurologici: Atassia spastica
- Atassia
- Spasticità
- Occasionali
- Atrofia ottica
- Epilessia
- Mentale disfunzione
- Polineuropatia: Pazienti gravi
- Patologia: Lieve perdita di assoni mielinizzati
- NCV: Lievemente rallentata
- MRI
- Leucoencefalopatia emisferica cerebrale diffusa
- Le aree di materia bianca anormale hanno il segnale T2 ad alta intensità
come il CSF
- Patologia
- Leucodistrofia ortocromatica cavitante senza atrofia: Nessuna placca
- Localizzazione: Materia bianca emisferica
- Oligodendrociti: Densità aumentata senza attività mitotica
- Astrociti: Riduzione; Riduzione del numero nelle regioni perivascolari
- Varianti
- Ovarioleucodistrofia: Mutazioni in eIF2B2, eIF2B4 e eIF2B5
- Ereditarietà dominante 28
- Epidemiologia: 1 famiglia; Madre e figlio
- Insorgenza adulta: 4a decade
- Sonnolenza, confusione mentale e emiparesi: Dopo URI con febbre
Leucoencefalopatia con coinvolgimento del tronco cerebrale e
del midollo spinale e innalzamento del lattato (LBSL) 33
l
Aspartil-tRNA sintetasi mitocondriale (DARS2)
; Cromosoma 1q25.1;
Recessiva
- Epidemiologia: Molteplici famiglie negli USA, Europa, Turchia e Russia
- Genetica
- Eterozigotica
- Tipo: Missenso o salto di esone al sito di splicing
- Proteina aspartile tRNA sintetasi mitocondriale
- Famiglia aminoacile tRNA sintetasi classe II
- Mutante: Attività ridotta
- Clinica
- Insorgenza: 3 ÷ 15 anni
- Atassia cerebellare: Lentamente progressiva; Gambe > braccia
- Spasticità: Gambe > braccia; Da lieve a grave
- Sensorio: Disfunzione della colonna dorsale del midollo spinale; Vibrazioni ridotte distalmente
- Cognitivo: Lieve danneggiamento e declino
- Muscoli: Nessun coinvolgimento preminente
- Laboratorio
- Anormalità nel segnale T2 alla MRI
- WM cerebrale: Specialmente corpo calloso posteriore
- Midollo spinale: Colonna dorsale del midollo spinale; Tratti corticospinali laterali
- Tratti troncocerebrali: Tratti piramidali; Lemnisco medio;
Peduncoli cerebellari
- Disomogeneo
- Lattato: Alto nella WM alla MRS; Normale nel siero
- Complessi mitocondriali nei fibroblasti: Normali
- NCV: Normale
- SSEP: Anormali
- CSF: Proteine e lattato normali
- Vedi anche
Malattia di Salla
l
Famiglia dei portatori di soluti 17, membro 5 (SLC17A5; Sialina)
; Cromosoma 6q14-q15;
Recessiva
- Mutazioni
- Finnica: Omozigotica Arg39Cys, Malattia più lieve
- Malattia grave: Altre delezioni, inserzioni, e missenso o nonsenso
- Proteina: Sialina
- Omologa ai trasportatori di glutamato e di fosfato
- Contiene 10 eliche transmembrana
- Proteina mutata: Traffico intracellulare attraverso il Golgi
anormale
- Malattia da accumulo di acido sialico
- Trasporto dell'acido sialico libero alla membrana
lisosomica: Ridotto
- Epidemiologia: Prevalente in Finlandia
- Clinica
- Insorgenza: 12 ÷ 18 mesi
- Atassia
- Ipotonia
- Ritardo mentale
- Epilessia (18%): Assenza; Età successiva
- Epatosplenomegalia
- Caratteristiche facciali grossolane
- Forme della malattia più gravi con grave ritardo mentale, attacchi
epilettici e ipotonia
- Decorso: Progressivo deterioramento nella 2a decade
- Sindrome variante
- Malattia infantile da accumulo di acido sialico (ISSD): Causata da mutazioni di delezione
- Laboratorio
- NCV: Polineuropatia demielinizzante (50%)6
- Pazienti: Gravemente disabilitati; I più anziani
- Ridotta velocità di conduzione nervosa
- Latenza distale prolungata
- Sialuria
- Linfociti: Vacuolizzati (4% ÷ 15%)
- Lisosomi: Ingrossati
- MRI
- Atrofia cerebellare progressiva
- Ipoplasia del corpo calloso
- Mielinizzazione ritardata
Atassia con paralisi degli adduttori laringei e neuropatia motoria 10
l
Recessiva
- Epidemiologia: Una famiglia italiana
- Insorgenza: 5a ÷ 6a decade; Disfonia
- Clinica
- Paralisi bilaterale degli abduttori laringei
- Atassia, cerebellare
- Andatura instabile
- EOM: Nistagmo; Inseguimento liscio anormale
- Dismetria degli arti
- Neuropatia motoria
- Debolezza: Distale; Gambe > braccia
- Cognizione: Normale
- Sensorio: Normale
- Laboratorio
- MRI: Atrofia cerebellare di verme ed emisferi
- Laringoscopia: Insufficiente abduzione delle corde vocali
- EMG: Denervazione distale e fascicolazioni
Atassia: Insorgenza adulta con lesioni talamiche 12
l
Recessiva
- Epidemiologia: Una famiglia Finnica
- Insorgenza
- 30 anni
- Disturbi dell'andatura
- Infanzia normale
- Clinica
- Cerebellare
- Atassia: Arti e andatura
- Disartria
- Nistagmo
- Disturbi dei nervi cranici
- Disfagia
- Paresi dello sguardo in alto
- Riflessi tendinei: Ridotti nelle gambe
- Sensazioni: Ridotto senso delle vibrazione e delle posizioni delle articolazioni
- CNS: Insufficienza cognitiva; Epilessia in 1 paziente
- Progressione: Su 1 ÷ 2 decenni
- Laboratorio
- MRI lesioni
- Talamiche (Simmetriche)
- Materia grigia del tronco cerebrale
- Materia bianca cerebellare
- Patologia
- Alterazioni vacuolari nel talamo
- Perdita neuronale nel midollo spinale, cervelletto e tronco cerebrale
- Conduzione nervosa: Neuropatia sensoria assonale
- SNAP: Assenti o ridotti nelle gambe
- Velocità di conduzione: Leggermente ridotta
Atassia con distrofia maculare ad occhio di bue 16
l
Recessiva
- Epidemiologia: Singola famiglia; Numerosi membri di una generazione
- Insorgenza: Disturbi dell'andatura; Fanciullezza
- Clinica
- Maculopatia: Atrofica; Configurazione ad occhio di bue; Acuità visiva ridotta
- Cerebellare: Disartria; Gambe
- Sensorio: Ridotto in piedi
- Riflessi tendinei: Vivaci
- Cognizione: Normale
- Laboratorio
- Scansione CT: Atrofia cerebellare, linea mediana
Atassia spinocerebellare con intrusioni saccadiche (SCASI; SCAR4)
l
Cromosoma 1p36; Recessiva
- Epidemiologia: Singola famiglia
- Insorgenza
- 3a ÷ 4a decade
- Disturbi dell'andatura
- Difficoltà di lettura
- Clinica
- Atassia: Andatura; Tronco; Arti; Disartria
- Occhio: Oscillazioni macrosaccadiche
- Orizzontali
- Le saccadi grandi hanno una maggiore velocità
- Indotte da ogni spostamento dello sguardo
- Presenza di intervalli intersaccadici
- EOM simili si possono avere nella atassia di Friedreich
- Anatomia: Le lesioni della linea mediana cerebellare coinvolgono il nucleo del fastigio
- Nessun nistagmo
- Tratto piramidale (100%)
- Riflessi tendinei aumentati
- Risposta dell'estensore plantare
- Miocloni (100%)
- Fascicolazioni (100%)
- Sensorio: Posizione delle articolazioni ridotta
- Piede cavo: Lieve
- Laboratorio
- Conduzione nervosa: Neuropatia assonale moto-sensoria
- EMG: Denervazione attiva distale nelle gambe
atassia spinocerebellare con movimenti degli occhi lenti (SDSEM)
l
Recessiva
- Epidemiologia: Famiglia araba di origine palestinese in Kuwait
- Clinica
- Atassia: Disturbi dell'andatura
- Movimenti degli occhi: Saccadici lente o assenti
- Scompenso intellettivo
- Disfunzione extrapiramidale
- Neuropatia periferica
- Anormalità scheletriche
- Decorso: Progressivo su 1 ÷ 2 decenni
- Patologia
- Tratti e nuclei del mesencefalo
Atassia spastica di Portneuf con leucoencefalopatia (ARSAL; SPAX3)21
l
Cromosoma 2q33–34; Recessiva
- Epidemiologia
- Geografia: Famiglie nella regione di Portneuf a sud di Quebec City, Canada
- Variabilità: Intra e inter famigliari
- Insorgenza
- Età: Media 8 anni; Gamma - Nascita ÷ 59 anni
- Motorio: Goffaggine; "Paralisi cerebrale"
- Clinica: Atassia spastica
- Cerebellare
- Atassia
- Disartria (74%)
- Nistagmo(44%): Orizzontale
- Motoneuroni superiori
- Spasticità
- Iperriflessia
- Stimolo urinario 57%
- Altre caratteristiche nei pazienti più gravi
- Scoliosi (35%)
- Distonia (57%)
- Insufficienza cognitiva (44%): Lievi
- Occhi: Atrofia ottica + Cataratta (9%): Due pazienti più anziani
- Insufficienza uditiva: Lieve (13%)
- Nervi periferici: Normali
- Gravità: Variabilità intrafamigliare
- MRI
- Atrofia cerebellare (100%): Verme e emisferi
- Nei pazienti più gravi
- Atrofia corticale (40%)
- Leucoencefalopatia (50%): T2 e FLAIR; Pazienti più anziani
Karak sindrome (NBIA2B)
l
Fosfolipasi A2, Gruppo VI (PLA2G6)
; Cromosoma 22q13.1;
Recessiva
- Epidemiologia: 1 Famiglia; Di Karak (Giordania del sud)
- Genetica
- Insorgenza: Precoce
- Clinica
- Atassia cerebellare: Progressiva
- Distonia
- Spasticità
- Declino intellettivo
- MRI: Segno "Occhio di tigre" segno
- Patologia: Deposito di ferro nel CNS
Atassia con paralisi dello sguardo verso l'alto 25
l
Recessiva
- Epidemiologia: Famiglia araba
- Insorgenza
- 3÷4 anni
- Tremore
- Disturbi dell'andatura
- Clinica
- Atassia: Arti; Andatura; Postura
- Disartria
- Attacchi epilettici: Mioclonici e tonico-clonici generalizzati,
- Movimenti degli occhi: Paralisi dello sguardo verso l'alto
- Riflessi
- Tendinei: Normali o ridotti
- Plantare: Estensore
- Neuropatia: Sensoria
- Ridotto senso delle vibrazione e delle posizioni delle articolazioni
- NCV: Perdita assonale
- Biopsia muscolare: Tipo raggruppante; Nessuna alterazione mitocondriale
- Cognizione: Normale
- Laboratorio
- EEG: Scariche epilettiformi
- MRI: CNS normale
SCAR8: Atassia cerebellare pura (ARCA1)32
l
Proteina nucleare di sviluppo sinaptico 1 (SYNE1)
; Cromosoma 6q25;
Recessiva
- Epidemiologia: Famiglie franco-canadesi
- Mutazioni
- Correlate al sito di splicing
- Sito accettore dello splice AG alla giunzione dell'esone 85 con
l'introne 84; 310067A-G
- L'introne 81: 306434A-G; Crea un nuovo sito criptico AG accettore
di porzione
- Terminazione prematura: R2906X; 334338–334342delATTTG; Q7640X
- Allelica con: MD di
Emery-Dreifuss di tipo 3
- Proteina Syne 1 proteina
- Ampiamente espressa
- Dimensione: Molto grande
- Localizzazione subcellulare:
Nucleare
- Contiene il dominio spectrina
- CNS
- Selettiva neuronale: Alta nelle cellule di Purkinje e nei
neuroni olivari cerebellari
- CPG2
- Variante di splice specifica cerebrale
della SYNE1
- Localizzata nella zona endocitica postsinaptica
delle sinapsi eccitatorie
- Distrugge l'internalizzazione dei recettori del glutamato
- Può essere necessaria per un rapido riciclo
dei recettori sinaptici del glutamato
- Giunzioni neuromuscolari: Ancora
i mionuclei specializzati sottostanti le NMJ
- Insorgenza
- Età: Media 30 anni; Gamma 17 ÷ 46 anni
- Segni: Atassia; Disartria
- Clinica
- Cerebellare
- Disartria (100%)
- Atassia degli arti (98%)
- Andatura atassica (98%)
- Oculare: Nistagmo, evocato dallo sguardo (13%); Saccadici anormali (31%);
Inseguimento oculare lento e a scatti (45%)
- Riflessi tendinei: Vivaci nel 26%
- Progressione: Lenta
- Laboratorio
- MRI: Atrofia cerebellare (100%)
- Muscoli: Pochi nuclei in prossimità delle giunzioni neuromuscolari; Normale la struttura della NMJ
- Sindrome simile: Atassia di Holmes
Atassia con epilessia e ritardo mentale 34
l
Cromosoma 16q21-23; Recessiva
- Epidemiologia: 1 Famiglia dell'Arabia Saudita, consanguinea
- Insorgenza
- Età: 9 ÷ 12 mesi
- Epilessia
- Clinica
- Epilessia
- Tipo: Generalizzata, Tonico-clonica
- Trattamento: Valproate; Fenobarbitone; Non utile in tutti
- Atassia
- Ritardo nel camminare
- Nistagmo
- Disartria
- Arti e tronco
- Ritardo mentale: Parlata a 2 ÷ 3 anni
- Riflessi tendinei: Ridotti
- Nessun mioclone o demenza
- Decorso: Lentamente progressivo
- Laboratorio
- MRI: Atrofia verme-cerebellare
- EMG: Normale
- Biopsia muscolare
- Dimensioni delle fibre muscolari: Variata
- Vacuoli: Irregolari, sarcotubulari; ? Artefatti
- EEG: Punte e onde lente
Ipoplasia cerebellare, ritardo mentale e
locomozione quadrupede (CAMRQ2)
l
Cromosoma 17p; Recessiva
- Epidemiologia: Famiglia turca di consanguinei di origine curda
- Clinica
- Cerebellare
- Disartria
- Dismetria
- Disdiadococinesi
- Incapacità di camminare bipedalmente: Locomozione quadrupede
- Ritardo mentale
- Può essere grave
- Assenza della parola
- Scheletrico
- Cifosi toracica
- Bassa statura
- Caratteristiche facciali: Grossolane
- Altre
- Laboratorio
- MRI
- Cervelletto: Ipogenesi e scissura nella linea mediana del verme
- Atrofia cerebrale
- Corpo calloso: Lieve ipoplasia
Atassia + ritardo mentale lieve e locomozione quadrupede (CAMRQ3)
50
l
Anidrasi carbonica VIII (CA8; CARP)
; Cromosoma 8q11-q12;
Recessiva
- Epidemiologia: Famiglia irachena
- Genetica
- Mutazione: Ser100Pro
- Stesso gene mutato del: Topo barcollante
- Proteina CA8
- Altamente espressa nelle cellule di Purkinje cerebellari
- Influenza il legame dell' inositolo trifosfato (ITP) al suo
recettore ITPR1 nel reticolo endoplasmatico
- Modula i segnali del calcio
- Clinica
- Insorgenza: Congenita
- Ritardo mentale
- Atassia
- Andatura: Quadrupede
Atassia + epilessia + disturbi
degli occhi 48
l
Collagene 18A1
; Cromosoma 21q22.3;
Recessiva
- Epidemiologia: Famiglia dell'India del nord, 2 sorelle affette
- Genetica
- Mutazione: Delezione di 2 paia di basi
- Allelica con: Sindrome di Knobloch
- Proteina COL18A1
- Catena α del collagene
di tipo XVIII
- Multiplessina: Proteina della matrice extracellulare che contiene molti
domini a
tripla elica
- Prodotta proteoliticamente dal frammento del terminale C del collagene di tipo XVIII:
Endostatina
- Clinica
- Età dell'insorgenza: 3 ÷ 12 anni
- CNS
- Atassia
- Braccia e gambe
- Disturbi dell'andatura
- Nistagmo: Sguardo laterale
- Ritardo intellettuale e declino cognitivo
- Attacchi epilettici: Generalizzati e mioclonici
- Occhi
- Glaucoma
- Dislocazione delle lenti
- Distrofia retinica e corneale
- MRI
- Polimicrogiria: Bilaterale, Frontale e temporale. C'è marcata
- Atrofia cerebellare
- Altre atrofie: Tronco cerebrale, Soprasensoriali, Midollo spinale cervicale
SeSAME
49
l
KCNJ10 (Kir4.1)
; Cromosoma 1q23.2;
Recessiva
- Nosologia
- SeSAME: Attacchi epilettici,
Sordità neurosensoria,
Atassia,
ritardo Mentale,
squilibrio
Elettrolitico
- EAST: Epilessia,
Atassia,
Sordità neurosensoria,
Tubulopatia
- Epidemiologia: Famiglie turche, britanniche, Canadesi e Afgane
- Genetica: Mutazioni
- Perdita di funzione
- Missenso o terminazione
- R65P; G77R; C140R; T164I; A167V; R297C
- Proteina KCNJ10 (Kir4.1)
- Canale K+ autocorreggente
- Espresso nella glia nel cervello e nel midollo spinale
- Ripristina il K+ rilasciato dalla ripolizzazione neuronale
- Probabilmente numerose mutazioni colpiscono l'attività del canale via alterata
interazione con PIP2
- Clinica
- Età dell'insorgenza: ≤ 5 anni
- Attacchi epilettici: Insorgenza 3÷4 anni; Tonico-cloniche
- Atassia: Tremore intenzionale; Disturbi dell'andatura
- Ritardo nello sviluppo: Ritardo mentale
- Perdita uditiva: Neurosensoria
- Laboratorio
- Squilibrio elettrolitico: Disturbo renale dei tubuli distali convoluti
- Ipopotassemia
- Alcalosi metabolica
- Ipomagnesemia
- Renina e aldosterone: Livelli elevati
- MRI
- Midollo spinale: Patologia della materia bianca
- Cervelletto: Piccolo, alcuni pazienti
- Nervi
- Ipomielinizzazione dei grossi assoni mielinizzati
- Perdita assonale
DISTURBI ATASSICI CONGENITI
Generale
- Aplasia cerebellare o vermale
- Solitamente recessiva
ATASSIE CONGENITE: AUTOSOMICA RECESSIVA o SPORADICA
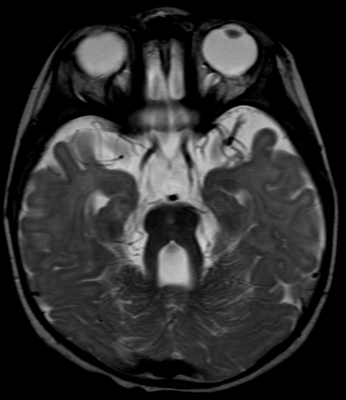
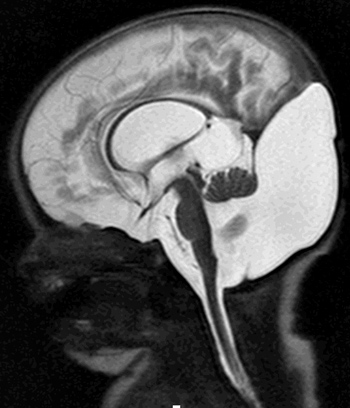
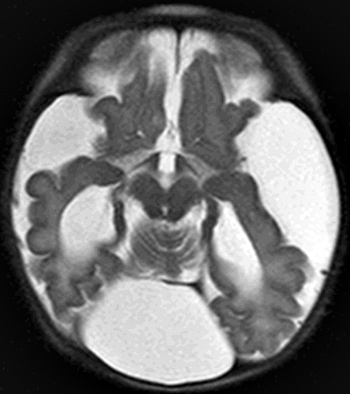
Sindromi di Joubert 26
- Sindromi di Joubert: Generale
52
- Mutato proteine: Ciliopatie
- Associato con la funzione del corpo ciglio/basale primario
- Altre ciliopatie
- Sindrome di Meckel (MKS)
- Sindrome di Bardet-Biedl (BBS)
- Nefronoftisi
- Amaurosi congenita di Leber (LCA)
- Caratteristiche
- Clinica e Laboratorio: Tutti i pazienti
- Insorgenza: Congenita
- Motorio: Ipotonia e ritardo nello sviluppo
- Atassia
- Disabilità intellettuale
- Disturbi del movimento degli occhi: Aprassia oculomotoria; Inseguimento liscio anormale;
Nistagmo
- Imaging: Malformazione mesencefalo–rombencefalo
- Ipoplasia del verme cerebellare
- Peduncoli cerebellari superiori: Ingrossati
- Segno del "dente molare"
- Altre caratteristiche comuni
- Encefalocele 6%: Specialmente nei tipi 5,
6, 7
- Disturbi retinici: Specialmente nei tipi 3,
5
- Coloboma corioretinico 19%
- Distrofia retinica 30%
- Malattia renale cistica 23%: Meno comune nei tipi
3, 8
- Malattia epatica: Ipertensione portale o altro 18%; Specialmente
nei tipi
5, 6,
7
- Polidattilia 19%: Specialmente nei tipi 6,
7
- Sindromi cliniche specifiche
- Cerebellare: Tipo 1
- Cerebellare + Corticale: Tipo 3
- CNS + multisistemica (Reni; Retina; Fegato): Tipo 2,
fenotipi variabili
- Collegamento esterno:
Segno del "dente molare"
- Sindrome di Joubert, tipo 1 (Disturbo cerebelloparenchimale IV; CPD IV)
l
Cromosoma 9q34.3; Recessiva
- Sindrome di Joubert di tipo A: Coinvolge cervello ma non i reni o la retina
- Genetica
- Insorgenza: Infantile
- Caratteristiche neurologiche
- Cerebellare: Atassia
- Occhi
- Oculomotorio: Nistagmo; Paresi dello sguardo verticale; Aprassia oculomotoria
- Ptosi
- Retinopatia: Distrofia; Coloboma; Retinite pigmentosa
- Ritardo mentale
- Protusioni ritmiche della lingua
- Respiratorio: Ipernea o apnea episodiche
- Caratteristiche sistemiche
- Cute: Fossette ai polsi e ai gomiti
- Scheletrico: Telecanto; Micrognatismo
- Malformazione cardiaca
- Patologia
- Aplasia vermale cerebellare
- Malformazioni del tronco cerebrale: "Segno del "dente molare""
- Profonda fossa interpeduncolare all'istmo e al ponte superiore
- Peduncoli cerebellari superiori allungati, ispessiti, e mal orientati
- Displasia del verme cerebellare superiore
- Assenza di decussazione dei peduncoli cerebellari superiori
- Mutazioni NPHP1: Il segno del "dente molare" è più leggero
- Sindrome di Joubert di tipo 2 (Sindrome cerebello-oculo-renale; CORS2)
19
l
TMEM216
; Cromosoma 11q13.1
- Sindrome di Joubert di tipo B: Coinvolge cervello, reni e retina
- Epidemiologia: Famiglie dal Pakistan, Emirati arabi uniti, Europa
- Genetica 54
- Mutazione negli ebrei ashkenaziti: R12L
- Insorgenza: Infantile
- Clinica: Variabilità fra famiglie
- Caratteristiche neurologiche
- Cerebellare: Atassia
- Idrocefalo
- Respiratoria: Ipernea o apnea episodiche
- Occhi
- Oculomotorio: Aprassia oculomotoria
- Insufficienza visiva
- Coloboma
- Renale
- Attraversati da molte piccole cisti
- Perdita della normale differenziazione cortico-midollare, bilaterale
- Dismorfismo facciale
- Ponte nasale depresso
- Ipertelorismo
- Palato ad arco alto
- Orecchie più in basso
- Patologia
- Aplasia vermale cerebellare
- Malformazioni del tronco cerebrale: "Segno del "dente molare""
- Corpo calloso: A gomito
- Sindrome di Joubert di tipo 3
l
AHI1; Cromosoma 6q23.2-23.3; Recessiva
- Epidemiologia: Famiglie turche e svizzere
- Genetica 22
- Mutazioni: Stop e missenso
- R351X; R435X; V443D
- Proteina AHI1
- Localizzazioni: Espressa nell'encefalo e nei reni
- Alti livelli negli assoni che non riescono ad attraversare linea mediana
nella sindrome di Joubert
- Insorgenza
- Età: Neonatale
- Problemi respiratori
- Ipotonia (100%)
- Clinica
- Cerebellare: Troncale (100%); Atassia (50%); Nistagmo (50%)
- Motorio: Tardi camminare o costretti in carrozzina
- Insufficienza cognitiva
- Visone ridotta
- Scheletrico: Cifoscoliosi (30%); Crescita ridotta (40%)
- Segno del "dente molare" (100%)
- Renale: Normale
- Laboratorio
- Verme cerebellare: Ipoplasia
- Alcuni pazienti hanno polimicrogiria
- Sindrome di Joubert di tipo 4
l
NPHP1
; Cromosoma 2q13;
Recessiva
- Epidemiologia: 2 fratelli e sorelle
- Clinica: Forma lieve della sindrome di Joubert
- Ritardo motorio: Lieve
- Oculomotorio: Aprassia; Capo inclinato
- Nefronoftisi
- MRI
- Verme cerebellare: Ipoplasia
- "Segno del "dente molare""
- Sindrome di Joubert di tipo 5
l
Proteina centrosomica, 290-KD (CEP290)
; Cromosoma
12q21.3; Recessiva
- Cervelletto-oculo-renale JSRD
- Epidemiologia: 8 famiglie
- Genetica
- Mutazioni: Missenso e terminazione
- Mutazioni anche nella
- Sindrome Senior-Loken 6
- Sindrome di Meckel
- Clinica: Forma variabile della sindrome di Joubert
- Ritardo motorio: Lieve; Ipotonia
- Atassia
- Oculomotorio: Aprassia; Capo inclinato
- Anormalità respiratorie neonatali
- Nefronoftisi
- Distrofia retinica
- Fenotipi incompleti: Sindromi cerebelloretiniche e cerebellorenali
- MRI
- Verme cerebellare: Ipoplasia
- "Segno del "dente molare""
- Sindrome di Joubert di tipo 6
l
Proteina transmembrana 67 (TMEM67; MKS3; Meckelina)
; Cromosoma
8q21.13-q22.1; Recessiva
- Epidemiologia: 4 pazienti
- Genetica
- Mutazioni
- Missenso, Sito di splicing e terminazione
- Eterozigotica
- Mutazioni anche in
- Insorgenza: Gestazione o fanciullezza
- Clinica
- Disabilità: Da lieve a grave
- Ipotonia
- Ritardo motorio
- Ritardo mentale
- Atassia cerebellare
- Movimenti stereotipati
- Anormalità respiratorie: Nei pazienti più gravi
- Coinvolgimento renale ed epatico: Alcuni pazienti
- MRI
- Verme cerebellare: Ipoplasia o displasia
- "Segno del "dente molare""
- Sindrome di Joubert di tipo 7
36
l
RPGRIP1-like proteina (RPGRIP1L)
; Cromosoma
16q12.2; Recessiva
- Cervelletto-renale JSRD
- Epidemiologia: 7 famiglie
- Genetica
- Mutazioni: Perdita di funzione e missenso
- Mutazioni presenti anche nei pazienti con la sindrome di Meckel:
Perdita completa della
funzione
- Proteina simile alla RPGRIP1
- Espressione: Ubiquitaria
- Localizzazione: Corpi basali alla base delle ciglia
- Interagisce con: Nefrocistina 4 (NPHP4)
- Clinica
- Atassia
- Ritardo nello sviluppo
- Movimenti degli occhi: Aprassia oculomotoria; Ptosi; Nistagmo
- Insufficienza renale: Nefronoftisi; Più tardi nell'infanzia
- Scheletrico: Scoliosi; Polidattilia
- Laboratorio
- MRI: Segno del "dente molare"
- Nessuna malattia retinica all'elettroretinogramma
- Sindrome di Joubert di tipo 8
41
l
Proteina simile al fattore di ribosilazione dell'ADP 13B (ARL13B; Proteina 1
simile a ARL2)
; Cromosoma 3q11;
Recessiva
- Proteina ARL13B
- Famiglia Ras GTPasi
- Funzioni: Cigliogenesi; Formazione dell'asse corporeo; Funzione renale
- Espressa nel cervelletto murino in sviluppo
- Localizzata al ciglio nei neuroni primari
- Clinica: Sindrome di Joubert tipica
- CNS
- Ipotonia
- Atassia
- Ritardo e ritardo psicomotorio
- Atassia oculomotoria
- Segno del "dente molare"
- Retinico: Normale
- Renale: Normale
- Topo: hennin
- Sindrome di Joubert di tipo 9 (JBTS9)
45
l
Proteina 2A contenente il dominio C2 a spirale avvolta (CC2D2A)
; Cromosoma
4p15.3; Recessiva
- Genetica: Mutazioni
- Perdita di funzione
- Omozigotiche: Spesso
- Missenso e nonsenso: P1122S; R1528C; R950X; L1551P; V1097FfsX1;
R1019X/pQ1096H
- Proteina CC2D2A
- Localizzata al corpo basale
- Co-localizza con cep290
- Espressa in tutti i tessuti fetale e adulti
- Clinica
- Sindrome di Joubert non complicata
- Movimenti oculari anormali: Comuni
- Distrofia retinica: Pochi pazienti
- Nessuna polidattilia
- Malattia renale ed epatica: Occasionali
- Alcuni pazienti con
- Laboratorio
- MRI: Segno del "dente molare"
- Sindrome di Joubert 10, collegata a X (JBTS10)
53
l
CXORF5 (OFD1)
; Cromosoma
Xp22.3-p22.2; Recessiva
- Famiglia della Malesia
- Genetica
- Mutazione frameshift : Esone 21; p.K948NfsX8, p.E923KfsX3
- Allelica con
- Sindrome orofaciodigitale 1 (OFD1)
: Collegata a X
dominante
- Sindrome di Simpson-Golabi-Behmel, Tipo 2 (SGBS2)
- Proteina
- Pericentriolo
- Si lega alla Lebercillina (LCA5)
- Formazione cigliare
- Clinica
- Sindrome di Joubert
- Ritardo mentale: Grave
- Movimenti oculari anormali: Comuni
- Retinite pigmentosa
- Polidattilia, postassiale
- Infezioni ricorrenti
- Apnea centrale
- Renale: Normale
- Decorso: Morte > 20 anni
- Laboratorio
- MRI: Segno del "dente molare"; Ipoplasia del verme cerebellare
congenita Atassie: Altre
- Sindrome COACH
l
Proteina 2A contenente il dominio C2 a spirale avvolta (CC2D2A)
; Cromosoma 4p15.3;
Recessiva
- COACH Nosologia: Ipoplasia del verme
Cerebellare, Oligofrenia,
Atassia, Coloboma,
Fibrosi epatica
- Genetica
- Caratteristiche neurologiche
- Atassia
- Insorgenza precoce
- Patologia: Aplasia vermale
- Ritardo mentale
- Spasticità
- Caratteristiche sistemiche
- Fibrocirrosi epatica
- Oculare: Coloboma, Ptosi
- Scheletro
- Ipertelorismo
- Faccia rotonda e piatta
- Polidattilia
l
Proteina transmembrana 67 (TMEM67; MKS3)
; Cromosoma
8q21.13-q22.1; Recessiva
- Genetica
- Composizione eterozigotica
- Frequenza delle mutazioni TMEM67 nella sindrome COACH: 57%47
- Le mutazioni TMEM67 causano anche: Sindrome di Joubert 6
- Clinica
- Sindrome di Joubert CNS
- Atassia
- Respirazione anormale
- Ritardo mentale e ritardo nello sviluppo
- Aprassia oculomotoria
- Fegato
- Epatomegalia
- Fibrosi epatica congenita
- Renale: Nefronoftisi
- Sindrome dell'andatura da dandy
l
ZIC1
e ZIC4
geni; Cromosoma 3q24-q25.1; Recessiva o sporadica
- Sindrome dell'andatura da dandy 251
l
FOXC1
; Cromosoma 6p25.3;
Famigliare o sporadica
- Genetica
- Mutazioni: Delezioni e duplicazioni al locus del cromosoma 6p25.3
- Le delezioni coinvolgono FOXC1 più almeno due esoni di GMDS
- Sindrome più grave comprendente DWS
- Grande delezioni coinvolgenti almeno sette geni (IRF4
÷ GMDS)
- Duplicazioni: Fenotipo più lieve; Andatura da dandy non completa
- Proteina Foxc1
- Ampiamente espressa nel mesenchima
- Partecipa ad una ampia gamma di processi di sviluppo
- Influenza la proliferazione e la differenziazione regolando
- La secrezione delle molecole di segnalazione: Bmp2, Bmp4, Cxcl12, Tgfb1
- Clinica: Vedi Andatura da dandy 3q24
- Patologia
- Malformazioni cerebellare e della fossa posteriore
- Ipoplasia del verme cerebellare
- Mutazione: Delezioni più piccole
- Clinica: Sindrome più lieve
- Megacisterna magna
- Malformazioni nella andatura da dandy
- Mutazione: Delezioni più grandi
- Clinica: Sindrome più grave
- Corpo calloso: Corto in alcuni pazienti
- Difetti meningei
- Occhi: Sindrome Axenfeld-Rieger
- Sindrome delle glicoproteine carenti di carboidrati: Tipo 1a (CDG1A)
l
Tipo 1a: Fosfomannomutasi 2 (PMM2)
; Cromosoma
16p13.2-p13.12; Recessiva
- Caratteristiche neurologiche: Più preminente nei pazienti più anziani
- Caratteristiche sistemiche: Presenti durante il primo periodo dall'insorgenza
- Capezzoli invertiti
- Occhi a mandola; Strabismo
- Degenerazione pigmentaria retinica
- Dita fusiformi
- Maggior preminenza delle labbra
- Lipodistrofia: Natiche
- Cardiaco: Cardiomiopatia ipertrofica; Effusioni pericardiache
- Disfunzione epatica
- Laboratorio
- Siero: Livelli ridotti di
- Glicoproteine
- Acetilglucosaminiltrasferasi N
- Glicoproteine: Livelli ridotti di
- Acido sialico, galattosio e acetilgalattosamina N
- Patologia
- Casi gravi con OPCA
- Ipoplasia cerebellare
- CDG: Altri tipi non-neurologici
- Tipo 1b: Fosfomannosio isomerasi-1
- Disturbo GI con perdita di proteine da enteropatia
- Trombosi e sanguinamento pericoloso per la vita
- Trattamento: Mannosio orale
- Tipo 4
- Clinica
- Microcefalia; Attacchi epilettici; Palato ad arco alto
- Siero
- Transferrina carente di carboidrati moderatamente elevata
- Agenesi cerebellare e pancreatica 24
l
Fattore di trascrizione pancreatico 1, subunità α (PTF1A)
; Cromosoma
10p13-p12.1; Recessiva
- Epidemiologia: Famiglie pakistane e nord europee
- Mutazioni: Troncamento; 705insG, C866T
- Proteina PTF1A
- Fattore di trascrizione bHLH
- Espressa in pancreas e cervelletto in sviluppo
- Insorgenza: Congenita
- Clinica
- Diabete: Neonatale
- Ritardo nella crescita: Intrauterina, Grave
- Faccia dismorfica
- Triangolare
- Piccola mento
- Orecchie in basso
- Contratture: Flessione; Braccia e gambe
- Grasso sottocutaneo: Ridotto
- Respirazione: Irregolare
- Nervo ottico: Ipoplasia
- Decorso: Morte precoce
- Laboratorio
- Iperglicemia
- Insulina e peptide C serici: Molto bassi
- Imaging del cervello: Agenesi del cervelletto e verme
- Pancreas: Assente
- Paralisi cerebrale atassica
l
Cromosoma 9p12-q12; Recessiva
- Epidemiologia: Famiglia asiatica
- Clinica
- Atassia cerebellare
- Disartria
- Disdiadococinesi
- Nistagmo orizzontale
- Andatura a gambe larghe
- Paralisi cerebrale
- Spasticità: 25%
- Intelligenza: Normale
- Sindrome di Gillespie
l
Geni del box 6 appaiati (PAX6); Cromosoma 11p13; Sporadica o dominante
- Mutazioni
- Eterozigotica
- Terminazione o sito di splicing
- Produce anche: Aniridia; Ipoplasia del nervo ottico; Coloboma
- Clinica
- Aniridia
- Pupille dilatate fisse
- I bordi pupilla iride mostrano angoli smerlati 'festonati'
- Atassia cerebellare
- Ipotonia
- Ritardo mentale
- Faccia: asimmetrica; Fronte alta, Ptosi, Strabismo, Ipertelorismo,
Ponte nasale depresso
- Decorso: Non progressivo
- Patologia: Ipoplasia cerebellare
- Aprosencefalia e disgenesi cerebrale
- Aprosencefalia
- Chiasma ottico e mesencefalo: Assenti
- Metencefalo: Scarsamente formato
- Cervelletto: Gravemente displastico
- Displasia cerebellotrigeminale dermica
(Sindrome di Gomez-Lopez-Hernandez)
l
Recessiva o sporadica
- Scheletrico
- Craniosinostosi
- Ipoplasia della mediofacciale
- Bassa statura
- Opacità della faccia corneale, alopecia dello scalpo, orecchie in basso
- Neurologico
- Atassia
- Anestesia trigeminale
- Ritardo mentale
- Patologia del CNS
- Anomalie del tronco cerebrale: Rombencefalosinapsi
- Fusione degli emisferi cerebellari
- Agenesi o ipogenesi del verme
- Fusione del nucleo dentato e dei peduncoli cerebellari superiori
- Sindrome di Behr
l
Autosomica recessiva
- ? Disturbo eterogeneo
- Visione
- Atrofia ottica: Bilaterale
- Difetti del campo visivo: Temporale; Incompleto
- Atassia
- Midollo spinale
- Andatura spastica
- Perdita sensoria: Colonna posteriore del midollo spinale
- Ritardo mentale
- Atrofia: Distale
- Riflessi tendinei: Ridotti in alcuni pazienti
- Decorso: Inizialmente progressione, poi statico
- Atassia cerebellare 1 (CLA1; Norman; CPD III; SCAR2)
: Vedi anche
ipoplasia cerebellare
l
Cromosoma 9q34-9qter; Recessiva
- Epidemiologia: Famiglia libanese
- Atassia cerebellare: Non progressiva
- Deficienza mentale
- Bassa statura
- ± Albinismo
- 1° Atrofia delle cellule granulari del cervelletto
- Atassia cerebellare 3 (CLA3; SCAR6)
l
Cromosoma 20q11-q13; Recessiva
- Epidemiologia: Famiglia norvegese
- Insorgenza: Infanzia
- Clinica
- Sviluppo
- Ritardo nel camminare
- Ipotonia
- Movimenti impacciati
- Lento sviluppo della parola
- Atassia cerebellare
- Non progressiva
- Andatura atassica
- Tremore intenzionale
- Nistagmo
- Motoneuroni superiori
- Riflessi tendinei: Vivaci
- Spasticità: Lieve; Occasionali
- Intelletto: Normale
- Scheletrico
- Bassa statura
- Piedi piatti
- Laboratorio
- Imaging del CNS: Atrofia del verme cerebellare
- Lissencefalia familiare con palatoschisi e ipoplasia cerebellare
- Clinica
- Insorgenza: Ritardo nella crescita prenatale
- Decorso: Morte in 1° settimana
- Palatoschisi
- Patologia
- Lissencefalia (l'encefalo appare liscio e senza circonvoluzioni)
- Corteccia cerebrale: Spessa; Disorganizzata
- Ipoplasia cerebellare: Grave
- Lissencefalia famigliare con ipoplasia cerebellare
7
l
RELN
; Cromosoma 7q22;
Recessiva
- Nosologia: Norman-Roberts
- Mutazioni: Sito di splicing; Troncamento di proteina
- Proteina RELN
- Grande proteina secretoria (338 kD)
- Funzione: Ruolo nella stratificazione dei neuroni nella corteccia cerebrale e
nel cervelletto
- Meccanismo di azione: Ligando per i ricettori delle lipoproteine
VLDLR e ApoER2
- Clinica
- Ipotonia
- Ritardo cognitivo
- Attacchi epilettici
- Oculare: Nistagmo; Miopia
- Linfedema: Congenito; Si può risolvere nel 1° o nel 2° anno
- MRI
- Lissencefalia: Moderate
- Ipoplasia cerebellare: Grave
- Modello di topo: Reeler
- Ipoplasia cerebellare con semplificazione girale cerebrale (CAMRQ1)
l
Recettore per le lipoproteine a densità molto bassa (VLDLR)
; Cromosoma 9p24;
Recessiva
- Epidemiologia: Popolazione Hutterita; Famiglia turca
- Nosologia
- "Sindrome della mancanza di equilibrio"
- Vedi anche: Locomozione quadrupede
- Mutazione: Delezione omozigotica di 199.163 bp
- Proteina VLDLR
- Recettore per la reelina
- Processi di segnalazione reelinici: Migrazione guidata dei neuroblasti nella corteccia cerebrale e nel cervelletto in sviluppo
- Clinica
- Atassia cerebellare
- Specialmente troncale
- Deambulazione ritardata
- Ritardo mentale: Moderato ÷ profondo
- Strabismo: La maggiore parte dei pazienti
- Piedi piatti: La maggiore parte dei pazienti
- Attacchi epilettici: 40%
- Bassa statura: 15%
- Decorso: Non progressivo
- Patologia
- Cervelletto: Ipoplasia dell'inferiore
- Cervello: Lieve semplificazione del girale
- Microcefalia con polimicrogiria e agenesi del corpo calloso 43
l
Eomesodermina, Xenopus, omologa della (EOMES; TBR2)
; Cromosoma
3p21.3-p21.2; Recessiva
- Epidemiologia: Famiglia marocchina consanguinea
- Genetica: Mutazione
- Omozigotiche
- Traslocazione bilanciata tra i cromosomi 3p e 10q: Silenzia il
trascritto EOMES
- Proteina EOMES
- Coinvolta nella divisione e/o migrazione neuronale
- Fattore di trascrizione
- Membro della famiglia del Box T
- Clinica
- Microcefalia: Microcefalia ad insorgenza prenatale
- Ritardo motorio con ipotonia: Grave
- Letalità precoce nella maggioranza: Morte a 15–18 mesi di età dovuta a sofferenza respiratoria dopo infezioni croniche
- Bambini sopravviventi: Persistenza di febbre e di infezioni ricorrenti
- Laboratorio: MRI
- Agenesi del corpo calloso
- Polimicrogiria, bilaterale
- Dilatazione ventricolare
- Cervelletto: Piccolo
Ponto-Cerebellare Ipoplasia
- Ipoplasia pontocerebellare con atrofia cerebrale progressiva (PCH2)
44
- Caratteristiche generali della PCH2
- Microcefalia progressiva dalla nascita
- Discinesia extrapiramidale: Corea
- Epilessia
- Sindromi genetiche specifiche
- PCH2A
l
TSEN54
; Cromosoma 17q25.3;
Recessiva
- PCH2B
l
TSEN2
; Cromosoma 3p25.2;
Recessiva
- PCH2C
l
TSEN34
; Cromosoma 19q13.4;
Recessiva
- Epidemiologia
- Genetica
- Mutazioni
- Omozigotiche o eterozigotiche
- Missenso o stop
- TSEN54: Ala307Ser, Q246X, Q343X e S93P; europei
- TSEN2: Tyr309Cys; Pakistani
- TSEN34: Arg58Trp; Turchi
- Mutazioni TSEN2 anche in: PCH4
- Proteina TSEN54
- Parte de tRNA splicciante il complesso delle endonucleasi
- Insorgenza
- Congenita
- Insonnia
- Suzione o deglutizione scarse
- Clinica
- Microcefalia: Progressiva
- Paresi spastica
- Discinesia extrapiramidale: Grave
- Insufficiente ad acquisire capacità volontarie
- Laboratorio
- Imaging del CNS
- Ipoplasia pontocerebellare: Pan-cerebellare; Ponte ventrale
- Atrofia cerebrale : Progressiva
- Patologia del CNS
- Perdita di neuroni nel sistema olivopontoneocerebellare
- Foglio cerebellare corto con scarsa ramificazione ("ipoplasia"):
Verme risparmiato
- Zone con contorni demarcati aree di perdita per l'intero spessore della
corteccia cerebrale
- Perdita segmentale dei nucleo dentato con isolette preservate e
alterazioni reattive
- Perdita segmentale del nucleo olivare inferiore con alterazioni reattive
- Perdita di nucleo pontino ventrale con assenza di fibre pontine traverse
vicine
- Risparmiate le cellule del corno anteriore spinale
- Ipoplasia pontocerebellare con microcefalia progressiva (CLAM; PCH3)
l
Cromosoma 7q11-q21; Recessiva
- Epidemiologia
- Insorgenza
- congenita
- Microcefalia
- Ipotonia
- Clinica
- Caratteristiche craniofacciali
- Capo: Microcefalia; Brachicefalia
- Occhi prominenti
- Orecchie in basso
- CNS
- Ritardo nello sviluppo: Grave
- Attacchi epilettici: 1° anno
- Ipotonia
- Riflessi tendinei: Vivaci
- Atrofia ottica: 1 paziente
- Nessuna caratteristica extrapiramidale
- Laboratorio: Imaging del CNS
- Tronco cerebrale: Piccolo
- Cervelletto: Atrofia degli emisferi e del verme, verme cerebellare piccolo
- Cervello
- Prominente solchi e ventricoli laterali
- Diminuzione del volume della materia bianca cerebrale
- Ipoplasia pontocerebellare 6 (PCH6)
37
l
Arginil-tRNA sintetasi mitocondriale (RARSL; RARS2)
; Cromosoma 6;
Recessiva
- Epidemiologia: Famiglia ebrea sefardita
- Mutazione
- Omozigotica
- Sito di splicing, IVS2+5 a->g
- Produce frame shift
- Insorgenza: Congenita
- Clinica: Encefalopatia; CNS solamente
- Ipotonia
- Suzione scarsa
- Episodi apneici
- Attacchi epilettici
- Microcefalia
- Spasticità
- Laboratorio
- Enzimi ossidativi mitocondriali: Riduzione variabile dell'attività dei complessi
- I, III e IV, o complesso I o IV solamente
- Livelli di mtDNA: Alti
- MRI: Atrofia progressiva
- Cervelletto
- Ponte
- Corteccia cerebrale
- Materia bianca
- EEG: Attività epilettica generalizzata
- Lattato: Alto nel siero o nel CSF
- Altri difetti nel tRNA mitocondriale(Arg)
- Mutazione del mtDNA: Mutazione a->g alla posizione 10438
- Insorgenza: Fanciullezza; 8 anni
- Encefalopatia progressiva
- Ritardo mentale: Moderato
- Nistagmo
- Parlata disartrica ritardata
- Goffaggine
- Ipoplasia pontocerebellare (PCH): Altre sindromi recessive
- PCH4
: Infantile fatale con
relativa preservazione di verme e anormalità nella fogliazione cerebellare
l
TSEN2
; Cromosoma 3p25.2;
Recessiva
- Epidemiologia: 1 famiglia spagnola, 3 fratelli e sorelle
- Allelica con: PCH2B
- Clinica
- Insorgenza: Neonatale
- Microcefalia
- Mioclone
- Ipertonia
- Patologia: perdita neuronica nell'oliva inferiore e nucleo pontino
- PCH5
: Verme ipocellulare
e attacchi epilettici fetali
- Epidemiologia: 1 famiglia, 3 fratelli e sorelle
- Clinica
- Insorgenza: Fetale
- Attacchi epilettici fetali
- Morte: Prima della nascita, 3° trimestre
- Patologia: Perdita di volume diffusa nel CNS, Più preminente nel
cervelletto e nel tronco cerebrale
- PCH associata con altre sindromi
- Distrofie muscolari congenite
- Lissencefalia
- Anormalità cromosomiche
- Difetti nella glicosilazione delle proteine
ATASSIA CONGENITA: COLLEGATA A X
- Atassia congenita collegata a X
l
Cromosoma Xq23
- Clinica
- Atassia: Deambulazione all'età 1 ÷ 7
anni
- Parlata ritardata e disartrica
- Oftalmoplegia esterna
- Riflessi tendinei: Vivaci
- Nessun ritardo mentale, spasticità o perdita sensoria
- Decorso: Non progressivo dopo l'infanzia
- Patologia: Ipoplasia degli emisferi cerebellari e del verme
- Atassia congenita collegata a X (SCAX1)4
l
Cromosoma Xp11.21-q21.3; Recessiva
- Clinica
- Insorgenza: Nascita
- Ipotonia
- Ritardo motorio
- Oculare: Movimenti oculari lenti
- Atassia cerebellare: Non progressiva
- Cognitivo: Normale o lieve ritardo
- MRI: Normale inizialmente; Atrofia cerebellare all'età di 3
anni
- Atassia congenita con extrapiramidale coinvolgimento (SCAX2)
l
Cromosoma X; Recessiva
- Epidemiologia: Singola famiglia
- Insorgenza precoce
- Clinica
- Atassia
- Spasticità
- Rigidità
- Morte precoce
- Atrofia cerebellare: Assenza delle cellule di Purkinje
- Sindrome atassia-sordità (SCAX3)
l
Cromosoma X; Recessiva
- Clinica
- Insorgenza: Infanzia
- Ipotonia
- Atassia
- Sordità: Neurosensoria
- Ritardo nello sviluppo
- Oculare: Esotropia; Atrofia ottica
- Decorso: Progressivo; Morte nell'infanzia
- Femmine eterozigote: Possono avere episodi di atassia
- Patologia
- Perdita neuronale e gliosi del nucleo dentato e dell'oliva inferiore
- Nucleo rosso, nucleo motorio dorsale del vago e processi uditivi
centrali gravemente colpiti
- Atassia congenita con grave ritardo mentale
5
l
Cromosoma Xq24-q27.3; Recessiva
- Ritardo mentale
- Epilessia
- Oftalmoplegia
- Dismorfologia craniofacciale
- Atrofia cerebellare
- Sindrome Hoyeraal-Hreidarsson
l
Discherina (DKC1); Cromosoma Xq28; Recessiva
- Neurale
- Ipoplasia cerebellare: Ridotta cellularità dei piani molecolare e granulare
- Microcefalia
- Pancitopenia: Insorgenza 5 mesi; Progressive per anni
- Discheratosi congenita
- Ritardo nella crescita
- Ritardo mentale con epilessia,
ingrossamento ventricolare rostrale 17
l
Oligofrenina 1 (OPHN1)
; Cromosoma Xq12;
Recessiva
- Epidemiologia
- Ritardo mentale + anomalie cerebellari: 12% delle famiglie
- Ritardo mentale collegato a X : 1% delle famiglie
- Genetica: Mutazioni con perdita di funzione
- Riarrangiamento cromosomico: Femmine
- Delezioni: Grandi o piccole; Frameshift
- Alcuni delezioni probabilmente mediate da
ripetizioni Alu
- Proteina oligofrenina
- Proteina attivante la Rho GTPasi
- Espressa nelle cellule neuronali e gliali
- Interagisce direttamente con i filamenti F dell'actina
- Regola le dinamiche del citoscheletro attraverso la modulazione della Rho GTPasi
- Specificamente coinvolta nella morfogenesi della spina dendritica
- Clinica
- Variabilità: Intrafamigliari e interfamigliari
- Ritardo mentale
- Da moderato a grave
- Può essere la sola caratteristica con alcune mutazioni nell'oligofrenina 1
- Epilessia: Generalizzata
- Scatti mioclonici e assenze nel 1° anno
- 6 anni: Attacchi epilettici tonico-clonici generalizzati
- Motorio
- Ipotonia (Neonatale)
- Ritardo
- Cerebellare
- Dismetria
- Adiadococinesia
- Oculomotorio: Nistagmo, strabismo, oftalmoplegia esterna
- Andatura atassica: Insolita
- Tremore intenzionale
- Occhi
- Strabismo
- Ridotta acuità dovuta ad errori rifrattivi
- Profondi
- Scheletro
- Dismorfismo facciale
- Mento prominente
- Ipotelorismo
- Occhi profondi
- Arcate sopraorbitali prominenti
- Naso lungo tubolare
- Solco sottonasale corto
- Labbro superiore sottile
- Macrocefalia
- Statura grande
- Ipogenitalismo maschile: Scroto ipoplastico; Microfallo
- Patologia del CNS
- Ventricolomegalia: Laterale
- Ipoplasia cerebellare: Vermiano; Mancante di lamine; Asimmetrica
- Nucleo caudato del capo: Ipoplasia
- Nessuna progressione
- Laboratorio
- EMG e NCV: Normali
- Chimica ematica: Normale
- Femmine portatrici: Strabismo; Difficoltà di apprendimento
- Cataratte, atassia, bassa statura e ritardo mentale (CASM)
31
l
Cromosoma Xpter-q13.1; Semi-Dominante
- Epidemiologia: Famiglia cinese
- Insorgenza: Nascita
- Clinica
- Cataratte
- Lenti completamente opache
- Congenita
- Bassa statura: Minore del 5° percentile
- Ritardo mentale: Da lieve a moderato
- Ipotonia
- Atassia
- Distribuzione: Arti e tronco
- Disartria
- Andatura atassica: Deambulazione limitata o nessuna
- Femmine eterozigote: Cataratta congenita
- Atassia congenita collegata a X (SCAX5)
38
l
Cromosoma Xq25-q27.1; Recessiva
- Epidemiologia: Famiglia americana di ascendenza norvegese
- Insorgenza: Neonatale
- Clinica
- Ipotonia: Neonatale grave
- Sviluppo motorio: Inizialmente ritardato
- Movimenti degli occhi: Lenti
- Atassia
- Tremore
- Nistagmo
- Disartria: 1° anno di vita
- Non progressivo: I sintomi migliorano con l'età
- Deambulazione senza supporto
- Intelligenza: Normale/al limite
- Laboratorio
- MRI: ipoplasia cerebellare, globale
- Ritardo mentale collegato a X con bassa statura, testicoli piccoli,
atrofia muscolare e tremore
l
Cullina 4B (CUL4B)
; Cromosoma Xq23;
Recessiva
- Nosologia: Sindrome di Cabezas
- Mutazioni: Missenso, Troncamento e stop
- Proteina CUL4B
- Subunità della ubiquitina ligasi
(E3)
- Restringe la possibilità di duplicazione del DNA: Previene l'aberrante reiniziazione della duplicazione del DNA
- Facilita la degradazione di CDT1
- Processo nel quale ligandi solubili nei grassi regolano la degradazione
selettiva delle proteine bersaglio
- Proteina cullina: Funzione
- Proteina adattatrice per serie di complessi ubiquitina-proteina ligasi che
regolano la degradazione delle proteine
- Clinica
- Peso alla nascita: Basso
- CNS
- IQ basso (gamma 29÷54)
- Linguaggio: Insufficiente o assente
- Durata dell'attenzione: Diminuita
- Iperattività
- Balzi d'umore
- Comportamento aggressivo
- Attacchi epilettici
- Scheletrico
- Morfologia
- Obesità: Troncale
- Labbro inferiore: Prominente
- Spazio tra 1° e 2° dito dei piedi
- Motorio
- Tremore fine: Aumentato dalle attività motorie
- Atrofia muscolare: Gambe inferiori
- Andatura: A gambe larghe
- Coordinazione motoria fine: Diminuita
- Endocrine
- Ipogonadismo: Testicoli (< 10%)
- Ginecomastia
- Pituitaria: Normale
- Fenotipo con malformazione del cervello e con microcefalia collegati a
X
42
l
Proteina chinasi serina dipendente da calcio/calmodulina (CASK)
; Cromosoma Xp11.4;
Recessiva
- Genetica: Mutazioni
- Eterozigotica
- Perdita di funzione
- Proteina CASK
- Famiglia della guanilato chinasi associata alla membrana (MAGUK)
- Proprietà della famiglia
- Indirizzata alle sinapsi neuronali
- Regola il traffico di indirizzamento e segnalazione dei canali ionici
- CASK
- ‘Pseudokinase’
- Funziona come parte di un grande complesso di segnalazione sia nei siti pre-
che
postsinaptici
- Trasferisce al nucleo
- Interagisce con box T specifico del cervello membro della famiglia TBR1
- Regola l'espressione del gene
- Clinica
- Microcefalia: Congenita; Grave
- Ritardo mentale
- Perdita uditiva: Neurosensoria
- Laboratorio
- MRI cerebrale: Riduzione del numero e complessità dei giri; Tronco cerebrale sottile;
Ipoplasia cerebellare
- Patologia: Lamine cerebrali e corticali anormali
Atassia a tarda insorgenza
Atassie metaboliche
ATASSIE COLLEGATE A X
- Vedi anche: Atassia congenita
- Anemia sideroblastica e atassia spinocerebellare (XLSA/A)
l
Trasportatore cassette 7 legante ad ATP(ABCB7; ABC7)
; Cromosoma Xq13
- Trasportatore ABC7
- Famiglia ABC: Media il trasferimento dei soluti dipendente dall'ATP
- Struttura
- 2 Domini transmembrana: Formano un poro nella membrana e determinano la specificità del substrato
- 2 domini citosolici leganti ATP: Accoppiano il legante all'ATP al movimento dei soluti
- Localizzazione: Membrana mitocondriale
interna
- Funzione: Omeostasi del ferro; Esporta dalla matrice nello spazio
intermembrane
- Alti geni ABC mutati nella adrenoleucodistrofia,
fibrosi cistica
- Atassia di Friedreich anche correlata ai disturbi dell'omeostasi del ferro
- Caratteristiche neurologiche
- Atassia: Insorgenza ad 1 anno; Non progressiva
- Segni dei tratti lunghi: Iperriflessia, Babinski
- Caratteristiche ematologiche
- Anemia: Lieve non richiedente trasfusione
- Elevati livelli di protoporfirina eritrocitica libera
- Nessun deposito eccessivo di ferro parenchimale
- Sideroblasti inanellati nel midollo osseo
- Femmine eterozigote: Possono avere lieve anemia, ma non atassia
- Sindrome di Arts
l
Fosforibosilpirofosfato sintetasi 1 (PRPS1)
; Cromosoma Xq22.3
- Genetica
- Mutazioni: Missenso; Gln133Pro, Leu152Pro
- Allelica con
- CMT X5
- Iperuricemia, ritardo mentale e sordità neurosensoria con superattività
della PRPS1
- Proteina PRPS1: Mutazioni
- Perdita di funzione
- Può causare insufficiente biosintesi delle purine
- Insorgenza: prima fanciullezza
- Clinica
- Motorio
- Ipotonia: Insorgenza precoce
- Tetraplegia flaccida e ariflessia: Successive
- Atassia
- Responsabilità per le infezioni: Specialmente URI
- Sordità
- Prognosi: La maggiore parte con morte < 5 anni
- Laboratorio
- Ipoxantina: Non rintracciabile in urine
- Patologia: Perdita di assoni mielinizzati nella colonna posteriore del midollo spinale
- Femmine portatrici: Insufficienza uditiva nella prima età adulta
- Atassia con tremore e declino cognitivo (FXTAS)
14
l
Gene del
X fragile e ritardo mentale (FMR1)
; Cromosoma Xq27.3
- Genetica: Mutazioni nel gene FMR1
- Espansioni CGG: Media 84; Gamma
69 ÷ 135 ripetizioni
- "Allele della premutazione"
- Frequenza nella popolazione: 1 su 259 femmine; 1 su 813 maschi
- FMR1 mRNA: Elevato 4x; ? Aumento della funzione di mutazione da elevati
livelli dei messaggi
- Allelica con: Sindrome del X fragile e ritardo mentale
con > 200 ripetizioni CGG
- Proteina FMR1
- Localizzazione: Dendriti
- Malattia
- Livelli: Basso-normali o lievemente diminuiti
- Risulta: Ridotti livelli del recettore AMPA (Internalizzazione aumentata )
Storia familiare
- Positiva per adulti sindrome del CNS nel 40%
- Comunemente positiva per la sindrome del X fragile
- Penetranza: Probabilmente più alta con l'aumentare dell'età
Insorgenza
- Maschi > 50 anni
- Tremore: Il più comune
- Difetti delle funzione esecutiva
Clinica
- CNS
- Cerebellare
- Tremore intenzionale: Azione (70%) > Riposo (10%); 3
÷ 5 Hz; Può essere
asimmetrico; Progressivo
- Atassia: Disartria; Gambe; Posturale e disturbi dell'andatura; Dismetria
- Nistagmo
- Cognitivo: Declino; Ridotta memoria a breve termine
- Psichiatrico: Labilità umorale; Ansietà; Comportamenti reclusivi
- Parkinsonismo: Lieve; Bradicinesia e rigidità
- Segni piramidali: Riflessi tendinei vivaci; Dita dei piedi piegate verso l'alto
- PNS: Polineuropatia (60%)40
- Genetica: Più preminente con maggiore lunghezza delle ripetizioni della
mutazione
- Età dell'insorgenza: Tarda età adulta; 6a e 7a decade
- Clinica
- Perdita sensoria: Vibrazioni e puntura; Piedi
- Autonomo: Impotenza (80%); Urinario (55%); Incontinenza intestinale (30%);
Sincope
- Riflessi tendinei: Ridotti distalmente
- NCV
- ampiezza dei CMAP: Piccola
- Velocità: Leggermente rallentata
- Sensorio: Piccola ampiezza degli SNAP
- Patologia: Inclusioni intranucleari
- Localizzazione: Cellule gangliali sensorie e autonome
- Simili a: Malattia con inclusioni intranucleari neuronali ialine (NIHID)
- Possono contenere: Lamina A/C; Espansioni-ripetizioni del FMR1 mRNA
Debolezza (30%): Prossimale nelle gambe
Altro: Malattia cardiaca (50%)
Decorso
- Progressiva su decenni
- Può diventare più rapida dopo anestesia generale
- Morte: Correlata all'età dell'insorgenza; Insufficienza cardiaca congestiva
Laboratorio
- Velocità di conduzione nervosa: Lievemente ridotta
- MRI
- Intensità del segnale T2 aumentata nei peduncoli mediali cerebellari e
nella materia bianca cerebellare adiacente
- Simmetrica
Patologia del CNS 14
- Atrofia cerebrale: Generalizzata
- Cervelletto: Perdita di cellule di Punkinie e torpedini
- Inclusioni
- Eosinofile
- Intranucleari
- Neuroni e astrociti: Specialmente nell'ippocampo
- Colorazione per ubiquitina
Diagnosi differenziale: Atrofia sistemica multipla
Femmine portatrici: Menopausa prematura in 20% delle portatrici di
espansioni di premutazione
Atassia-demenza (SCAX4)
- Epidemiologia: Una famiglia
- Insorgenza
- Età: 2 ÷ 3 anni
- Ritardo nel camminare
- Tremore
- Clinica
- Atassia: Progressiva
- Segni del tratto piramidale
- Demenza: Insorgenza adulta; Problemi di memoria
- Morte: 6a decade
Ritardo mentale, microcefalia, epilessia e atassia
l
Famiglia dei portatori di soluti 9, isoforma A6 (SLC9A6)
; Cromosoma Xq26.3
- Nosologia
- Sindrome simile alla Angelman
- Tipo Christianson
- Genetica
- Mutazioni: Delezione; Stop; Sito di splicing
- Proteina SLC9A6
- Clinica
- Neonatale periodo: Normale
- Capo
- Microcefalia
- Crescita lenta
- Epilessia
- Età dell'insorgenza: 9 ÷ 26 mesi
- Molteplici tipi
- Atassia: Troncale; Andatura instabile
- Oftalmoplegia
- Ritardo mentale
- Grave
- Ritardo nello sviluppo
- Assenza della parola
- Spettro dell'autismo
- Episodi di riso non provocato
- Segni piramidali: Alcuni pazienti
- Riflessi tendinei: Vivaci
- Riflessi plantari: Estensore
- Spasticità
- Emiparesi
- Extrapiramidale e Movimenti
- Distonia
- Movimenti ipercinetici
- Mano strizzante
- Incontinenza
- Gastrointestinale
- Bocca: Aperta; Salivazione profusa
- Difficoltà di deglutizione
- Rigurgitazione frequente: Dovuta al riflusso gastroesofageo
- Morfologia: Sottile
- Laboratorio
- EEG: Attività epilettiforme; Retroterra di ritmo veloce
- MRI: Atrofia cerebellare, progressiva
- Patologia
- Perdita neuronica: Generalizzata
- Deposito di Tau
- Neuronale e gliale
- Modello simile alla degenerazione corticobasale
Piruvato
deidrogenasi: Atassia episodica
Pelizaeus-Merzbacher variante allelica:
Sindrome del PLP nullo
Atassia congenita collegata a X
Atassia: Disturbi multisistemici
Disturbi dei topi
- Reeler
 Informazioni per i pazienti
Informazioni per i pazienti
Ritorna a
Indice delle atassie
Ritorna a
Neuromuscolare Home Page
Riferimenti
1. Neurology 2001;56:849-855, Neurology 2003;60:1206–1208,
Neurology 2004;62:818–820,
Neurology 2007;68:295-297
2. J Med Genet 2000;37:1-8,
Pediatr Neurol
2003;28:335-341, J Neurol 2009;256 (Supp 1):3-8
3. Neurology 2000;54:1408-1414, Arch Neurol 2003;60:982-988
4. Am J Med Genet 2000;92:53-56
5. Med Genet 1999;36:759-766
6. Neurology. 2000;55:99-104.
7. Nature Genetica 2000;26:93-96
8. Am J Hum Genet 2000;67:1320-1326, Nature Genetica 2004; March
9. Am J Hum Genet 2001;68:501-508, Arch Neurol 2001;58:173-174, Hum Mol Genet
2004 Online March
10. Neurology 2001;57:1412-1414
11. Neurogenetics 2001;3:127-132
12. Neurology 2001;57:1043-1049
13. cuore 2002;87:346-349
14. Neurology 2001;57:127-130, cervello 2002;125:1760-1771, Am J Hum Genet
2003;72:869–878
15. J Clin Endocrinol Metab, aprile 2002:87:1607–1612,
Pediatr Neurol
2010;42:359-364
16. Am J Ophthalmol 2002:133:410-413
17. cervello 2003;126 agosto, J Med Genet 2003;40:441–446
18. Neurology 2003;61:274-275
19. Am J Hum Genet 2003; Online agosto
20. Nature Genet 2003; Online ottobre
21. cervello 2006; On-line maggio 3
22. Nature Genet 2004; Online agosto
23. Nature Genet 2004; Online settembre
24. Nature Genet 2004; Online novembre
25. Neurology 2005;64:142–144
26. Ann Neurol 2005;57:513–519
27.
Paediatr Anaesth 2005;15:433-434
28.
Ann Neurol 2005 Jul 26
29.
Hum Mol Genet 2005; Online Aug
30. Nature Genetica 2005; Online Nov 13
31.
J Hum Genet 2006; Jul 11
32.
Nat Genet 2006 Dec 10
33.
Nat Genet 2007;39:534-539
34.
cervello 2007; Online aprile
35.
Neurology 2004;62:818-820
36. Nature Genetica 2007; Online June
A;
B
37.
Am J Umani Genet 2007; Online luglio
38. Am J Med Genet A 2008
Feb 1
39. Am J Umani Genet
2008;82:623–630,
2008;82:661–672
40. Am J Med Genet Parte A
143A:2256–2260
41. Am J Umani Genet 2008;
Online Aug
42. Nature Genet 2008; Online agosto
43. Nature Genet 2007;39:454-456
44. Nat Genet 2008 Aug 17
45. Am J Hum Genet 2008
Oct 22
46. American Journal of
Umani Genetica 2008; novembre
47. Hum Mutat 2008 Dec 4
48. American Journal of
Medica Genetica 2009 Jan 21
49. Proc Natl Acad Sci U S
A 2009 Mar 16,
NEJM 2009;360:1960-1970
50. PLoS Genet 2009
maggio;5(5):e1000487
51.
Nature Genet 2009; Online agosto
52. Semin Pediatr Neurol
2009;16:143–154
53. American
Journal of Umani Genetica 2009; ottobre
54. Am J Hum
Genet 2009; Online Dec
20 aprile 2010
Va a
Acronimi e sigle
Va a
Mitolario in italiano
Va a
Mitolario
italiano - inglese
Va a Mitolario
inglese - italiano