![]() Traduzione di Natale
Marzari
Traduzione di Natale
Marzari
Home,
Ricerca,
Indice alfabetico,
Collegamenti,
Patologia,
Molecole,
Sindromi,
Muscoli,
Giunzioni neuromuscolari, Nervi, Spinale, Atassia, Anticorpi e Biopsia, Informazioni per i pazienti

| Fondazione FONAMA | fonama@fonama.org | Telefono 335250742 |
|
Giunzioni neuromuscolari, Nervi, Spinale, Atassia, Anticorpi e Biopsia, Informazioni per i pazienti |
|
|
SLA ereditarie e famigliari Dominanti SLA 1: SOD1; 21q SLA 3: 18q21 SLA 4: Senatassina; 9q34 SLA 6: FUS; 16p11 SLA 7: 20p13 SLA 8: VAPB; 20q13 SLA 9: Angiogenina; 14q11 SLA 10: TDP 43; 1p36 SLA 11: FIG4; 6q21 SLA: DAO; 12q24 SLA-FTD 1: 9q21-q22 SLA-FTD 2: 9p21 SLA bulbare Dinactina: 2p13 Recessive SLA 2: Alsina; 2q33 SLA 5: Spatacsina; 15q21 SLA 6: FUS; 16p11 SLA 6-21: 6p25, 21q22 Collegata a X SLA X: Xp11-q12 Altre (Sporadiche) Neurofilamenti, subunità pesante: 22q12 Periferina: 12q12 SLA del Pacifico occidentale-PD1 SLA-PD2 SLA-FTD-3: CHMP2B; 2p11; Sporadica Insorgenza nella fanciullezza HMN distale: 7q34; Dominante SLA2: Alsina; 2q33; Recessiva SLA4: Senatassina; 9q34; Dominante SLA5: Spatacsina; 15q21; SLA recessiva 6-21: 6p25, 21q22 Altre (Tipo 2) Sindromi bulbari AAA, sindrome: Aladina; 12q13; Recessiva Brown-Vialetto-van Laere: c20orf54; 20p13; Recessiva BSMA: Dominante Fazio-Londe: Recessiva o dominante Kennedy, sindrome (BSMA): Recettore degli androgeni; Xq12 PLS, giovanile: Alsina; 2q33; SLA recessiva bulbare Worster-Drought Disturbi multisistemici AAA, sindrome: Aladina; 12q13; Recessiva ANE: RBM28; 7q31 Camera-Marugo-Cohen, sindrome Cataratte e anormalità scheletriche: Dominante DDPAC: MAPT; 17q21; Dominante Esoaminidasi A: Esoaminidasi β A; 15q23; Recessiva Machado-Joseph: Atassina-3; 14q32; Dominante Malattia con corpi poliglucosanici: GBE1; 3p12; Recessiva Mitocondriali: SCO2 MND + demenza e oftalmoplegia: Recessiva Paraparesi spastica + neuropatia motoria Ritardo mentale + atrofia distale: CUL4B; Xq23 |
Atrofia muscolare spinale (SMA): Tipi SMA1: SMN 5q; Recessiva Congenita con artrogriposi Kugelberg-Welander Werdnig-Hoffmann SMA recessive: Altre Atrofia muscolare spinale 2 (SMA2) Mitocondriali SCO2: 22q13 TK2: 16q22 SMA + fratture congenite SMA + ipoplasia pontocerebellare: VRK1; 14q32 SMA collegate a X SMA (Recessiva) SMA bulbare (di Kennedy): Recettore degli androgeni; Xq12 SMA e artrogriposi infantili (SMAX2): UBE1; Xp11 SMA distale, collegata a X (SMAX3): ATP7A; Xq13 SMA prossimali dominanti Benigna congenita con contratture Congenita con debolezza delle gambe: 12q23; Dominante HMSN-P (Okinawa tipo): 3q13; Dominante Insorgenza adulta: VAPB; 20q13 Sindrome scapoloperonea: 12q24 e altro SMA bulbare autosomica dominante HMN e SMA distale 1: 7q34; Dominante 2A: HSPB8 (HSP22); 12q24; Dominante 2B: HSPB1 (HSP27); 7q11; Dominante 2C: HSPB3 (HSPL27); 5q11; Dominante 5: Predominanza degli arti superiori HMN 5A: GARS; 7p15; Dominante HMN 5B: BSCL2; 11q13; Dominante 6: IGHMBP2; 11q13; Recessiva 7: + Paralisi delle corde vocali HMN 7A: 2q14; Dominante HMN 7B: Dinactina; 2p13; Dominante + Motoneuroni superiori: Senatassina; 9q34; Dominante HMN: 11p; Recessiva HMN: 16p SMA distale (DSMA) 1: IGHMBP2; 11q13; Recessiva 2 (HMN J): 9p21.1-p12; Recessiva 3: 11q13; Recessiva 4 (Sindrome dei motoneuroni inferiori): PLEKHG5; 1p36 Collegata a X (SMAX3): ATP7A; Xq13 Predominante nelle gambe + atassia telangectasia: ATM; 11q22 Insorgenza nella fanciullezza Dominante Recessiva Distale ulnare-mediano: Dominante Contratture congenite letali Scapoloperoneale Vedi anche Collegamento esterno: Database sulle mutazioni SLA Amiotrofia monomelica (Hirayama): Sporadica Modelli di topo |
|
SMA: Caratteristiche cliniche Congenite Artrogriposi SMA 0 Tipi: 1; 2; 3; 4 Motoneuroni inferiori SMA: Clinica - Correlazioni genetiche SMA: Esame genetico SMA: Patologia SMN geni: SMN1; SMN2 Proteina SMN Altre correlate e geni vicini 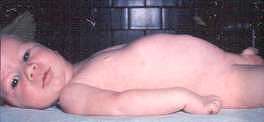 Di: Andrew Kornberg MD |
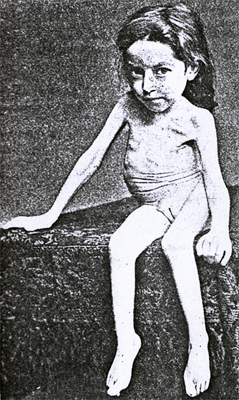 Hoffman ~1891 |
e viene rapidamente degradato
|

|
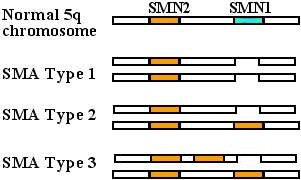
CROMOSOMI
5q Tipica SMN mutazioni in SMA |
|---|
|
|
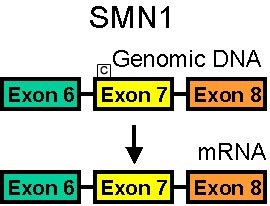
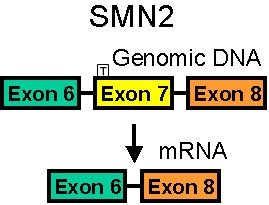
SMN1 Gene normale
Tipi di mutazione SMN1 Delezione Conversione a gene SMN2 Gene SMN2 Più copie: Correlate con SMA più lieve. Mutazioni SMN2 da sole: Non producono la SMA |
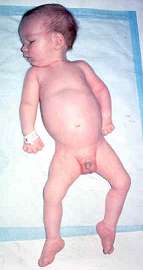 Da: A Kornberg MD |
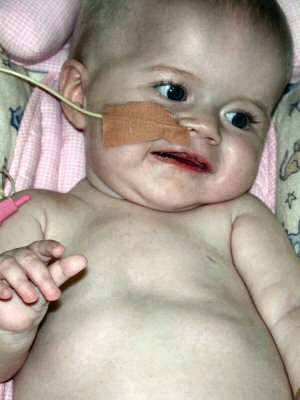 Da: M Ryan MD |
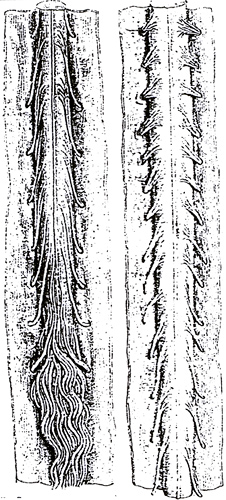 Midollo spinale nella SMA
Le radici anteriori sono atrofiche |
|
Proteina recettore degli androgeni Caratteristiche cliniche Correlazioni genetico-cliniche Epidemiologia Caratteristiche di laboratorio Insorgenza Meccanismi patogeni Patologia |
|
|
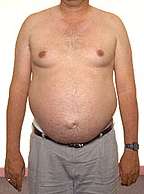
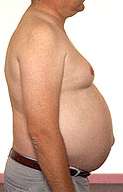
Atrofia muscolare bulbo-spinale Ginecomastia |

Bocca nella BSMA
tendente al sorriso e al riposo |

|
|
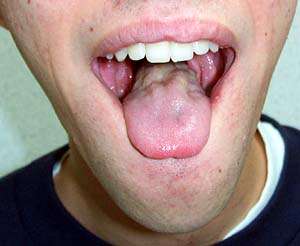
Lingua nella BSMA Atrofizzata; Debole; Si muove rapidamente |
|
HMN e SMA distale 1: 7q34; Dominante 2A: HSPB8 (HSP22); 12q24; Dominante 2B: HSPB1 (HSP27); 7q11; Dominante 2C: HSPB3 (HSPL27); 5q11; Dominante 5: Predominanza degli arti superiori HMN 5A: GARS; 7p15; Dominante HMN 5B: BSCL2; 11q13; Dominante 6: IGHMBP2; 11q13; Recessiva 7: + Paralisi delle corde vocali HMN 7A: 2q14; Dominante HMN 7B: Dinactina; 2p13; Dominante + Motoneuroni superiori: SETX; 9q34; Dominante HMN: 11p; Recessiva HMN: 16p |
SMA distale (DSMA) 1: IGHMBP2; 11q13; Recessiva 2 (HMN J): 9p21.1-p12; Recessiva 3: 11q13; Recessiva 4 (sindrome LMN): PLEKHG5; 1p36 Collegata a X (SMAX 3): ATP7A; Xq13 Gambe predominante + atassia telangectasia: ATM; 11q22 Insorgenza nella fanciullezza Dominante Recessiva Ulnare-mediano: Dominante Scapoloperonea Vedi anche SMA congenita (12q23) Hirayama |
Classificazione di Harding Tipo 1: AD; Insorgenza 2 ÷ 20 anni Tipo 2: AD; Insorgenza 20 ÷ 40 anni Tipo 3: AR; Lievi; Insorgenza 2 ÷ 10 anni Tipo 4: AR; Grave; Insorgenza 0,3 ÷ 20 anni Tipo 5: AD o sporadica; Arti superiori; Insorgenza 5 ÷ 20 anni Tipo 6: AR; Grave infantile Tipo 7: AD; Corde vocali; Insorgenza 10 ÷ 20 anni |
|
Ereditarietà Dominanti SLA 1: SOD1; 21q SLA 3: 18q21 SLA 4: Senatassina; 9q34 SLA 6: FUS; 16p11 SLA 7: 20p SLA 8: VAPB; 20q13 SLA: TDP 43; 1p36 SLA 9: Angiogenina; 14q11 SLA 11: FIG4; 6q21 SLA: DAO; 12q24 SLA-FTD 1: 9q21-q22 SLA-FTD 2: 9p21 SLA bulbare Dinactina: 2p13 Recessive SLA 2: Alsina; 2q33 SLA 5: Spatacsina; 15q21 Collegata a X SLA X: Xp11-q12 SLA sporadica-FTD-3: CHMP2B; 2p11 Neurofilamenti catena pesante: 22q12 Periferina: 12q12 Pacifico occidentale |
Età dell'insorgenza Insorgenza adulta SLA 1: SOD1; 21q SLA 3: 18q21 SLA 6: FUS; 16p11 SLA 7: 20p13 SLA 8: VAPB; 20q13 SLA: TDP 43; 1p36 SLA 9: Angiogenina; 14q11 SLA 11: FIG4; 6q21 SLA: DAO; 12q24 SLA X: Xp11-q12 SLA Altre: Gruppo eterogeneo SLA-FTD 1: 9q21-q22 SLA-FTD 2: 9p21 SLA-FTD-3: CHMP2B; 2p11 SLA bulbare Neurofilamenti catena pesante: 22q12 Dinactina: 2p13 Pacifico occidentale Insorgenza nella fanciullezza (giovanile) SLA 2: 2q33; SLA recessiva 4: Senatassina; 9q34; Dominante SLA 5: Spatacsina; 15q21; Recessiva Altre (Tipo 2) |
|
Collegamento esterno:
Database sulle mutazioni SLA | |
| Caratteristiche | SLA ereditaria | SLA sporadica |
| Maschi:femmine | 1:1 | 1,7:1 |
| Durata della malattia | Bimodale < 2 e > 5 anni |
Unimodale 3÷4 anni |
| % dei casi SLA | 10% | 90% |
| Insorgenza | ||
| Distribuzione per età | Più più giovani | Più anziani |
| Età media | 46 anni | 56 ÷ 63 anni |
| Giovanile | SLA 2, 4, 5 | Rara |
| Caratteristiche bulbari | 20% ÷ 30% | 10% ÷ 20% |
| Gambe | Comune | Occasionali |
|
SLA 1: SOD1; 21q SLA 3: 18q21 SLA 4: Senatassina; 9q34 SLA 6: FUS; 16p11 SLA 7: 20p13 SLA 8: VAPB; 20q13 SLA 9: Angiogenina; 14q11 SLA 10: TDP 43; 1p36 SLA 11: FIG4; 6q21 SLA: DAO; 12q24 SLA-FTD 1: 9q21-q22 SLA-FTD 2: 9p21 SLA X: Xp11-q12 SLA bulbare Dinactina: 2p13 |
|
Caratteristiche cliniche Generale Sindromi specifiche Mutazioni Localizzazione Aspetti funzionali Correlazioni specifiche Patologia Statistiche di popolazione Altro sulla SOD Proteina SOD |
| Sindromi SLA : Correlazioni con le specifica mutazioni SOD | |
|---|---|
|
l
Esone 1; Ala4Val La mutazione più comune Rapida insorgenza e progressione (1,0 anni) Frequentemente segni solamente nei motoneuroni inferiori l Esone 2; His46Arg @ Cu al sito di legame della SOD Insorgenza successiva; Nelle gambe Bulbare insolita Progressione lenta (17 anni) l Esone 2; Delezione di 6 bp (ΔG27/P28) 65 La mutazione riduce la trascrizione Bassi livelli della proteina SOD1 mutante Fondatore filippino Bassa penetranza Durata della malattia: 4,3 anni l Esone 4; Leu84Val Motoneuroni inferiori solamente Progressione rapida (18 mesi) ? Insorgenza più precoce in maschi l Esone 4; Asp90Ala Insorgenza: 20 ÷ 94 anni; Gambe; Fase preparetica Crampi alle gambe; Mialgie; Parestesie dolorose Disfunzione della vescica Progressione: Lenta; Gambe ® Braccia Ereditarietà Recessiva: Finnica (2,5% portatori) Dominante: Altri pazienti l Esone 4; Ile104Phe Caratteristiche cliniche intrafamigliari variabili Età dell'insorgenza: 6 anni - asintomatica Decorso: 2 ÷ 14 anni fino ai segni bulbari Insorgenza negli arti: Braccia o gambe l Esone 4; Ile113Thr Riportato in pazienti con la SLA sporadica Relativamente comune; Bassa penetranza Tarda Insorgenza: Media 59 anni Decorso: Variabile; 2 ÷ 20 anni l Esone 5; Codone 126 Delezione di 2 paia di basi Progressione rapida |
l
Caratteristiche generali della sindrome
|
 Inclusione di conglomerato ialino |
|
SLA2: 2q33 SLA5: Spatacsina; 15q21 SLA 6: FUS; 16p11 SLA 6-21: 6p25, 21q22 |
|
Brown-Vialetto-van Laere SLA bulbare Fazio-Londe Sindrome di Kennedy Madras malattia dei motoneuroni Atrofia muscolare spino-bulbare : Dominante Worster-Drought |
|
Va a Acronimi e sigle
Va a Mitolario italiano - inglese
Va a Mitolario inglese - italiano
|
Ritorno a Fonama.org Home Page |
Alla pagina originale
|
|