Traduzione di Natale
Marzari
Traduzione di Natale
Marzari
Home,
Ricerca,
Indice alfabetico,
Collegamenti,
Patologia,
Molecole,
Sindromi,
Muscoli,
Giunzioni neuromuscolari,
Nervi,
Spinale,
Atassia,
Anticorpi e Biopsia,
Informazioni per i
pazienti
|
 |
FIBRE MUSCOLARI ATTIVITA' e CRAMPI
Miotonia
Caratteristiche generali della miotonia
-
Clinica: Ritardato rilassamento dei muscoli scheletrici
dopo una contrazione volontaria
-
Classificazione
per malattie
dei canali ionici
-
Malattie
miotoniche da canale Na+: Miotonia aggravata da
potassio (PAM)
-
Fluttuazioni nel tempo negli individui:
Comune
-
Variazione fra le famiglie: Comune; Da giorno a giorno
-
Famiglie PAM più comuni della miotonia congenita
dominante (DMC)
mediata Cl-
-
Sindromi: Dominanti
-
Malattie
miotoniche da canale Cl-
-
Fluttuazione nel tempo negli individui: Insolita ma può accadere
-
Variazione tra le famiglie: Insolita; Durata
da settimane a mesi se presente
-
Sindromi
-
? Altra causa
-
EMG: Miotonia
-
Elettrofisiologia: Aumentata eccitabilità delle fibre muscolari
-
Potenziali di azione ripetitivi in
singole fibre muscolari
-
Avviene in risposta a stimolazione meccanica o elettrica
-
Sensibilità alla temperatura
-
Frequenze
-
Variabili
-
Spesso decrescenti nel tempo: 50 Hz ® 10 Hz
-
Ampiezza: Può variare fino a 10 fold
-
Durata
-
Corse miotoniche: Più lunghe nella distrofia miotonica che nella
miotonia congenita
-
Distrofia miotonica: Molte corse > 2 sec e
la più lunga di 30 sec
-
Miotonia congenita: La maggior parte corse < 1 sec e la più lunga di è 10 sec
-
Miotonia:
Rispondente alla mexiletina 15
-
Effetto del farmaco: Blocco dei canali Na+
-
Efficace nel trattamento della miotonia
-
Risposta migliore quando il farmaco blocca i canali mutanti
voltaggio-dipendenti
-
Qualche effetto in casi gravi con il bloccaggio dei canali di tipo selvatico
-
Derivati da
Me7: Più efficaci della mexiletina; Inibisce i canali Na+ in
modo controllato da M2
-
Mexiletina non efficace nel trattamento della paralisi periodica iperkalemica
Miotonia congenita (Thomsen)

l
Canali cloro muscolari (CLCN1)
;
Cromosoma 7q35; Dominante
-
Caratteristiche genetiche: Le mutazioni
-
Solo dominanti: Gly200Arg; Ile290Met; R317Q; Pro480Leu

-
Dominante e recessive: Gly230Glu; Ala313Thr; Arg317Gln;
Arg338Gln; Ile556Asn; Arg894X
-
Altro: S132C, L283F, T310M, T550M
-
Correlazioni
fisiologia-genetica
-
Canali
Cl- normali
-
Formate da un complesso tetramero
-
La conduttanza dei
Cl- è necessaria per stabilizzare il potenziale di membrana
-
Le mutazioni nei canali
Cl-
-
Interferisce con la formazione del dimero e del tetramero: Effetti negativi dominanti
-
La conduttanza dei
Cl- attraverso i canali è eliminata o ridotta
-
Le mutazioni frame deriva: Completa perdita delle proteine di canale
-
Missenso: Numerosi meccanismi di funzione patologica del canale
-
Spostamento del voltaggio verso valori positivi al di sopra dei quali avviene il
controllo: Gly200Arg
-
Probabilità di apertura del canale vicina a 0 al
potenziale di riposo
-
Cambiamenti nella selettività di canale
-
Rapporto di permeabilità catione:anione
più alto che nel tipo selvatico: G230E
-
Alterazioni nelle funzioni di canale
dipendenti dal voltaggio o da [Cl-]in
-
S132C
-
Canali omodimerici: [Cl-]in-dipendenti dalla attivazione
indotta dalla iperpolarizzazione
-
Canali eterodimerici: Attivazione indotta da depolarizzazione con attivazione
spostata (nel tempo ndt)
-
L283F
-
Difetto di
deattivazione ad alto [Cl-]in
-
T310M
-
Curva di attivazione: Spostata al normale [Cl-]in; Normale ad alto [Cl-]in
-
Senza la conduttanza dei Cl-: ± Aumenta la conduttanza cationica
-
K+ si accumula normalmente nei tubuli t dopo i potenziali di azione
-
Ma questo produce depolarizzazione della membrana 10x > normale
-
Maggiore depolarizzazione se il canale mutante è permeabile ai cationi
-
Questa post-depolarizzazione aumentata può raggiungere la soglia
-
Risultano spontanei inneschi dei potenziali di azione
-
L'integrità dei tubuli t richiede una scarica miotonica
-
Le mutazioni con fenotipi dominanti e recessivi
-
? Espressione più bassa dell'allele mutante nelle famiglie recessive
-
Caratteristiche cliniche
-
Insorgenza: Infanzia ad adulti
-
Penetranza variabile: 90% sintomatiche
-
Miotonia
-
Solitamente lieve
-
Decorso: Spesso costante; Può essere variabile nel tempo
-
90% alla EMG; 50% percussione
-
Variabilità
-
Esacerbazioni: Miotonia può peggiorare con freddo o lo sforzo
-
Fluttuazioni a lungo termine nella miotonia: Alcune mutazioni (Gly200Arg)
-
Assenza di sintomi per settimane o mesi
-
Sintomi aumentati dal freddo, fame, fatica e stato emotivo
-
Miotonia e debolezza in gravidanza: Alcune mutazioni (Gly230Glu; T310M)
-
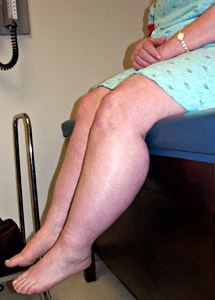
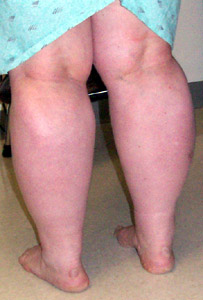
Ipertrofia muscolare
-
Mialgie: In alcuni pazienti
-
Fenomeno del
"riscaldamento" (miotonia ridotta dopo
attività ripetuta)
-
Forza muscolare
-
Normale a riposo in molti pazienti
-
Muscoli prossimali: Difficoltà funzionali (salire le scale) o lieve debolezza
permanente in alcuni
-
Debolezza distale può aversi con l'esercizio
-
Laboratorio
-
CK: Leggermente elevato o normale
-
EMG
-
Miotonia
-
Presente persino nella prima fanciullezza
-
Distale > prossimale
-
Effetto riscaldamento: Miotonia ridotta dopo un periodo di contrazione massimale
-
Potenziali delle unità motorie: Normali, non miopatia
-
Stimolazione ripetitiva del nervo
-
Decremento
-
Specialmente con stimolazione ad alte frequenze (30 Hz)
-
Presente con alcune mutazioni (P480L; R894X) e non altre22
-
Non facilitazione
intratetanica
-
Nessun effetto con il raffreddamento
-
Farmacologia:
-
Aggravata da
-
β2-agomisti: Fenoterolo; Ritodrina
-
Amino acidi
monocarbossilici
-
Rilassanti
muscolari
depolarizzanti
-
Trattata da
-
Chinina (200 a 1,200 mg/d)
-
Mexiletina (150 a 1,000 mg/giorno)
-
Dilantina (300 a 400 mg/giorno; Livelli terapeutici di 10 a 20 μl/mL)
-
Procainamide (125 a 1,000 mg/giorno)
-
Tocainide (soppressione del midollo osseo è un effetto collaterale)
-
Carbamazepina
-
Acetazolamide ( 125 a 750 mg/giorno)
Miotonia congenita (Becker)
l
Canali cloro muscolari (CLCN1)
; Cromosoma 7q35;
Recessiva
-
Caratteristiche genetiche
-
La maggior parte delle mutazioni CLCN1 produce sindromi recessive
-
Le mutazioni puntiformi
-
Molte mutazioni missenso: > 20 identificate
-
Localizzazione: Dal terminale N al dominio transmembranico XII
-
Altre mutazioni
-
Delezioni: Nucleotidi 1282-1285 e 1437-1450
-
Nonsensi: Q74stop; E193X
-
Sito di splicing nucleotide 979
-
Composizione eterozigote: Molti pazienti; Alcuni omozigoti
-
Topo (mutazioni recessive)
-
Sviluppo arrestato alla risposta richiesta (ADR):
-
Inserzione
di transposone
ETn
-
Miotoniche (MTO)
-
Fisiologia: Attivazione corrente alla iperpolarizzazione vs. disattivazione nel tipo selvatico
-
Non o ridotta apertura al voltaggio positivo del potenziale di equilibrio del Cl-: M485V
-
Spostamento
a destra (a maggiore potenziale +) del voltaggio dipendente della
probabilità di apertura dei canali ClC-1: I329T; R338Q
-
Rettificazione retrograda permette efflusso di Cl- ma non afflusso
-
Non identificati meccanismi fisiologici per alcune mutazioni: Y150C; Y261C
-
Caratteristiche cliniche
-
Insorgenza: 4 a 12 anni; Gambe
-
Miotonia
-
Con fenomeno "riscaldamento"
-
Più grave che nella forma dominante (Thomsen)
-
Ipertrofia muscolare
-
Specialmente gambe e glutei
-
? Maschi > femmine
-
Forza
-
Inizialmente normale
-
Rapido decremento con brevi esercizi
-
Ritorna normale con ulteriori contrazioni muscolari
-
± Lieve debolezza distale
-
Portatori: Miotonia in EMG dei maschi ma non delle femminile
-
EMG
-
Miotonia
-
Distale > Prossimale
-
Effetto riscaldamento: Meno miotonia dopo contrazione massimale
-
Unità motorie: Lievemente miopatiche
-
Decrementale risposta CMAP
-
Specialmente: A stimolazione ad alta frequenza (30 Hz); A seguito di esercizio
-
Nessun effetto del raffreddamento
Miotonia levior
l
Canali cloro muscolari (CLCN1); Cromosoma 7q35; Dominante
-
Mutazione: Gln552Arg
-
Caratteristiche cliniche
-
Miotonia: Lieve
-
Insorgenza: Tarda
-
Non ipertrofia muscolare
l
Canale del sodio - subunità α (SCN4A)
;
Cromosoma 17q35; Dominante
l
Canali cloro muscolari (CLCN1)
; Cromosoma 7q35; Dominante
-
Definizioni
- Paramiotonia: Miotonia "anormale"
- Miotonia paradossa: Miotonia peggiorante con l'esercizio ripetitivo
- Genetica
- Localizzazione delle mutazioni SCN4A
- Spire citoplasmatiche fra le ripetizioni I e II, e III e IV (inattivazione
della porta)
- Segmenti transmembranici S2, S3 e S4
- Specialmente nel lato citoplasmatico della membrana
- Correlate alla paramiotonia
- "Quarto cavallo": Mutazione Phe-a-Leu nel dominio transmembranico IVS-3
- Mutazione CLCN1: F428S
- Elettrofisiologia: Effetti della mutazione
SCN4A
- Difetti funzionali nei canali Na+: Anormale inattivazione; Minore
dipendenza dal voltaggio
- Ostacolano l'inattivazione rapida del canale Na+
- Gli ioni Na+ continuano a mancare dentro la cellula
- Alcune mutazioni ostacolano anche l'inattivazione lenta del canale Na+
- Inattivazione è prolungata dallo stato di canale aperto, ma normale
nello stato chiuso
- Conduttanza di canale: Permeazione di ioni Na+ attraverso il poro è normale
- Scarsa inattivazione del canale Na+ : Conseguenza elettrofisiologiche
- Potenziali di azione
prolungati
- Ridotta ripolarizzazione della membrana
- Correlazioni cliniche
- Lieve depolarizzazione (> 5mV) produce scariche ripetitive (miotonia)
-
Depolarizzazione più grave (> 20mV) produce debolezza
- Caratteristiche cliniche: Debolezza o rigidità possono aversi da sole
- Miotonia: Dovuta a depolarizzazione della membrana
- Causata da lieve rallentamento dell'inattivazione del canale Na
- Peggiora con freddo e esercizio
- Specialmente in faccia, collo e estremità superiori
- Insorgenza nell'infanzia
- Debolezza episodica: Dovuta al bloccaggio dei potenziali di azione
muscolare
- Causata da lieve rallentamento dell'inattivazione del canale Na
- Associata con paralisi periodica iper K+
- Dopo esercizio, esposizione al freddo, o spontanea
- Solitamente dura pochi minuti, ma occasionalmente giorni
- Insorgenza nell'adolescenza o mai
- Alcuni pazienti peggio dopo carico di K+
- Esacerbata dall'ipertiroidismo
- Clinica delle sindromi di "miotonia"
- Miotonia aggravata dal potassio (PAM)
- Attacchi precipitati dal carico di K+
- Non debolezza
- Sindromi includono:
- Le mutazioni SCN4A
Ser804Phe; Ile1160Val; Gly1306Ala;
Gly1306Val; Gly 1306Glu; Val1589Met
- Paramiotonia congenita (PMC)
- "Debolezza" dovuta a paramiotonia precipitata da ipokalemia
- Le mutazioni SCN4A
Val1293Ile; Thr1313Met; Leu1433Arg; Arg1448Cys;
Arg1448His; Arg1448Pro; G1456E; Val1458Phe; Phe1473Ser
-
Le mutazioni
CLCN1 18
- F428S: Missenso
- Canali: Normale fisiologia; Bassa espressione
- Paramiotonia congenita senza paralisi da freddo: Val1293Ile
- Epidemiologia: Famiglia irlandese
- Insorgenza
- Fanciullezza
- Spasmi dei muscoli irrigiditi: Prevalentemente colpite mani e
faccia
- Clinica
- Miotonia
- Peggiora con l'esercizio (miotonia paradossa)
- Esacerbazione più marcata: Tempi freddi
- Non paralisi periodica o debolezza progressiva
- EMG
- Profuse scariche miotoniche
- Raffreddamento: 78% di diminuzione dell'ampiezza CMAP
-
EMG: Miotonia
- Meno pronunciata che nelle altre miotonie: Può essere assente alla
normale temperatura
- Potenziali di azione
- Frequenza: 20 a 50 Hz
- Lunghezza dei treni d'onda: > 1 minuto
- Effetti del raffreddamento
- Iniziale: Scariche ripetitive spontanee delle unità motorie
- Aumentando il raffreddamento: Depolarizzazione e paralisi
® la miotonia scompare
- Terapia
- Miotonia: Farmaci
antiaritmici
- Mexiletina 150 a 1.000 mg/giorno
- Bloccaggio dei canali sodio dipendente dall'uso
- Riduce le serie ripetitive dei potenziali di azione
®
Ridotta rigidità muscolare
- ? Tocainide
- Debolezza: Sensibile al K+
- Idroclorotiazide (250 a 1.000 mg/giorno)
- ± Integrazione di K+
- Acetazolamide (125 a 1.000 mg/d)
- Diclorfenamide (50 a 150 mg/giorno)
- Debolezza: Paramiotonia
- Mexiletina 150 a 1.000 mg/giorno
- ? Tocainide
- K+
Miotonia fluttuante
l Canale del sodio - subunità α (SCN4A)
; Cromosoma 17q35; Dominante
-
Caratteristiche cliniche
-
Lieve miotonia che varia in gravità da giorno a giorno
-
Non debolezza o sensibilità
al freddo
-
Rigidità si sviluppa durante riposo 1/2 ore dopo esercizio; permane per ~1 ora
-
Rigidità esacerbate da
-
Potassio
-
Agenti
depolarizzanti (per es. Suxametonio)
-
Può interferire con la respirazione
-
EMG
-
Miotonia: Aumentata dopo l'esercizio
Miotonia permanente
l Canale del sodio - subunità α (SCN4A)
; Cromosoma 17q35; Dominante
l
Mutazione nella regione codificante il cardine del controllo dell'inattivazione del canale (Gly1306Glu)
-
Caratteristiche cliniche
-
Miotonia continua grave (Può interferire con la respirazione)
-
Ipertrofia muscolare marcata
Miotonia congenita
rispondente all'acetazolamide
l Canale sodio - subunità α (Ile1160Val) (SCN4A)
; Cromosoma 17q35; Dominante
-
Caratteristiche cliniche
-
Ipertrofia muscolare
-
Miotonia
"Paradossa": Peggiora con
attività ripetuta
-
Dolore muscolare e rigidità
-
Aggravata da potassio
-
Migliora con acetazolamide
-
Non debolezza
Distrofia miotonica (DM1)
l
Proteina
miotonina chinasi; Cromosoma 19; Dominante
Miopatia
motonica prossimale (PROMM; DM2)
l
Proteina a dita di zinco 9 (ZNF9)
;
Cromosoma 3q21; Dominante
PROMM
l
Loci non collegati a 3q21; Autosomica Dominante
-
Famiglia tedesca
-
Miotonia
-
Debolezza; Prossimale
-
Catarrate
-
MRI: Alterazioni nella materia bianca
Miotonia
acquisita : Farmacotossicità
Paralisi periodica iperkalemica
l
Canale sodio - subunità α (SCN4A)
;
Cromosoma 17q35; Dominante
-
Mutazioni4
-
Localizzazioni: Thr704Met; Ser906Thr; Ala1156Thr; Met1360Val; R1448C; Met1592Val
-
Meccanismi di azione: Difettosa inattivazione del canale Na+
-
Correlazioni clinico-genetiche
-
Thr704Met
-
Debolezza permanente comune
-
La mutazione più frequente: 60% dei casi HRPP
-
Met1592Val
-
Miotonica
-
Non distrofica
-
Frequenza: 30% dei casi HRPP
-
Ala1156Thr e Met1360Val: Ridotta penetranza
-
Altre mutazioni SCN4A nelle
-
Fisiologia: Le mutazioni con "esaltazione" delle funzioni"
|
Meccanismi della debolezza episodica |
|---|
|
Incremento del K+ extracellulare: 2° all'assunzione di K+, o
al riposo dopo esercizio
ê
Depolarizzazione delle membrane: Lieve
ê
Apertura dei canali Na+:
Anormali canali Na+ in modalità non-inattivabile
ê
é
Persistente corrente di Na+ verso l'interno
é
ê
é
í
Incremento del K+ extracellulare
Sostenuta depolarizzazione delle membrane muscolari
î
é
ê
efflusso
di
K+
Inattivazione dei canali Na+ normali
ê
Perdita della eccitabilità elettrica delle membrane muscolari
ê
Debolezza
|
-
Caratteristiche cliniche
-
Debolezza episodica: Flaccida
-
Insorgenza: 1a decade
-
Distribuzione
-
Solitamente prossimale e simmetrica
-
Sporadicamente distale e asimmetrica nei muscoli esercitati
-
Più frequente con l'aumento dell'età
-
Insorgenza
-
Prima di colazione
-
Durante il riposo dopo l'esercizio
-
Possono essere associate con parestesie
-
Durata 15 a 60 minuti: Sporadicamente ore o giorni
-
Provocate da
|
Esercizio
Assunzione di potassio
Ambienti freddi
Gravidanza |
Glicocorticoidi
Stress
Etanolo
Digiuno |
-
Attenuate da: Assunzione di carboidrati; Esercizio lieve
-
Prognosi: Debolezza
-
Può diventare permanente con alcune mutazioni SCN4A (Thr704Met)
-
Altre (Met1592Val) solo episodica
-
Comparazione degli attacchi alla
paralisi periodica ipopotassica
-
Frequenza: Maggiore
-
Durata: Più breve
-
Miotonia ± Paramiotonia
-
Episodica
-
Presente fra episodi di debolezza
-
Frequenza: Nella maggioranza dei pazienti con EMG
-
Rigidità al raffreddamento meno comune che nella paramiotonia
-
Disritmie cardiache: Escludere la sindrome di Andersen
-
Laboratorio
-
Attacchi associata ad alto K+ serico (>4.5 mEq/l) e
alto K+
urinario
-
CK serico: Normale a 300 U/L
-
Ampiezza
CMAP
-
Aumentati immediatamente dopo contrazione massimale sostenuta (5 min)
-
Progressivamente ridotti (del 40%) durante il riposo 20 a 40 min dopo iniziale incremento (80% dei pazienti)
-
Normali: Lievi aumenti delle ampiezze CMAP dopo esercizio
-
Esami provocativi
-
Carico orale di K+
-
Solo se le funzioni renale e cardiaca e il K+ serico sono normali
-
Eseguire al mattino a digiuno, dopo esercizio
-
0,05 g/kg KCl in liquido senza zuccheri in 3 minuti
-
Se negativo si può arrivare da 0,10 a 0,15 g/kg KCl
-
Monitorare gli elettroliti, ECG e forza ogni 20 minuti
-
Debolezza tipicamente maggiore da 90 ai 180 minuti
dopo
-
Esercizio
-
Cicloergometro
-
30 minuti
-
Pulsazioni da 120 a 160
-
Quindi riposo assoluto a letto
-
Misurare il K+ serico
-
Normale: Crescita del K+ durante l'esercizio; Poi cade vicino
ai livelli
pre-esercizio
-
Iperpotassemia PP: 10 a 20 minuti dopo esercizio ® 2o periodo iperpotassico e
paralisi
-
? Varianti: Paralisi periodica
normopotassica
-
Malattie animali: Quarter Cavalli
-
Genetica
-
Prole dell'americano Quarter Horse sire, Impressive
-
Mutazione
Phe1421Leu nel dominio transmembranico IVS-3
-
Clinica
-
Debolezza può essere grave
-
Ipertrofia muscolare
-
Omozigoti più gravi degli eterozigoti: Disfunzione
laringea e faringea
-
Trattamento
-
Molte attacchi brevi e non necessitano di trattamento
-
Attacchi acuti: Ingestione di
carboidrati
-
Cronica: Acetazolamide; Diuretici tiazidi
-
Non di beneficio per debolezza: Mexiletina
-
Esaminazione genetica:
Bambini di Washington
l
Sindrome di Andersen:
Paralisi periodica sensibile al K+ con disritmie cardiache e caratteristiche dismorfiche
Sindromi
dei muscoli increspati 17
Sindromi dei muscoli increspati: Ereditarie, Dominante
l
RMD1
:
Cromosoma 1q41; Dominante
l
RMD2
: Caveolina-3
;
Cromosoma 3p25; Dominante
l
? Altri loci; Dominante9
-
Genetica
-
Le mutazioni caveolina 3 : Missenso
-
R26Q: AD-RMD; IperCKemia
-
Asp27Glu: AD-RMD; Debolezza distale
-
A45T: AD-RMD e IperCKemia con sofferenza muscolare
-
Ala45Val: AD-RMD
-
P104L: AD-RMD e LGMD1C
-
Omozigote L86P e A92T: Malattia dei muscoli increspati,
grave
-
Le mutazioni in caveolina-3 causano anche
-
Caratteristiche cliniche
-
Epidemiologia
-
Femmine meno coinvolte dei maschi
-
Variabile all'interno delle famiglie
-
Insorgenza
-
Solita: 1a o 2a decade
-
Gamma: 5 a 54 anni
-
15% asintomatiche
-
Sofferenza
-
Crampi, dolore e rigidità
muscolari
-
Gambe > braccia
-
Rigidità del collo in alcuni pazienti
-
Peggiora con: Esercizio; Sporadicamente freddo
-
Dita camminanti: Al risveglio o dopo il riposo
-
Increspature dei muscoli: Simili a onde
-
Evocate dallo stiramento muscolare
-
Si muovono lateralmente lungo i muscoli per 5 a 20 secondi (0.6 M/s)
-
10x più lente dei potenziali di azione delle fibre muscolari
-
Elettricamente silenti
-
Presente nel 60% dei pazienti
-
Localizzazione: Prominenti specialmente nei grossi muscoli prossimali
-
Rilievo muscolare (Mioedema)
-
Evocato dalla percussione: Tutti i pazienti
-
Rapido
-
Diagnosi differenziale: Malnutrizione; Cachessia; Ipotiroidismo
-
Contrazione
-
Evocata da: Percussione muscolare (Lieve); Pressione
-
Ampiezza aumenta con la forza della percussione
-
Non abituazione
-
Caratteristica comune: Fino ai 100%
-
Insorgenza rapida
-
Durata: Fino a 30 secondi
-
Dolorosa nel 30%
-
Distribuzione: Generalizzata o in muscoli isolati
-
Clinicamente simile alla miotonia o mioedema
-
Ipertrofia muscolare: Polpacci o
generalizzata
-
Debolezza: Alcune mutazioni (Asp27Glu)
-
Prossimale o distale
-
Scapole alate
-
Possono essere esacerbate dal dantrolene
-
Sistemico
-
Strabismo: Intermittente
-
Pigmenturia: Ricorrente
-
Cardiomiopatia: Alcuni tipi genetici
-
Laboratorio
-
EMG
-
Attività muscolare è elettricamente silente
-
Alcuni aumenti dell'attività inserzionale
-
CK serico
-
Solitamente alto: Fino a 10x
-
Sporadicamente normale
-
Patologia muscolare
-
Dimensione delle fibre muscolari: Varia
-
Ipertrofia delle fibre di tipo 2
-
Nuclei centrali
-
Ridotta colorazione disferlina, specialmente con composizione mutazionale eterozigote
-
Colorazione caveolina: Ridotta
-
Ultrastrutture: Alterazioni più gravi con mutazioni omozigote
-
Completa assenza del caveolae nel sarcolemma
-
Plasmalemma: Discontinuità; Proiezioni papillari
-
Vacuoli subsarcolemmali
-
Fisiologia muscolare: Alterata regolazione Ca++ nel reticolo sarcoplasmatico
-
"Post-contrazione" della singole fibre muscolari
-
Alterato rilascio o apporto del Ca++ nel reticolo sarcoplasmatico
-
Trattamento
-
Dantrolene: Può aumentare la debolezza
-
Benzodiazepine
Sindromi dei muscoli increspati: Ereditarie, Recessive 14
l
Recessive
Sindrome
Schwartz-Jampel (miotonia condrodistrofica)
24
l
Perlecan
;
Cromosoma 1p34-p36; Recessive
- Genetica
- Le mutazioni
- Troncamento (inserzioni o delezioni) il più comune
- Missenso o splicing in alcuni pazienti
- Le mutazioni nulle nella
- Displasia
dissegmentale tipo Silverman-Handmaker
- Perlecan
-
Proteoglicano solfato di eparan
- Maggiore componente delle membrana basali e della matrice interstiziale
nelle cartilagini
- Funzioni
- Corecettore per
FGF2
- Interagisce con i componenti della membrana basale ed i recettori sulla superficie cellulare
- Integrina
β1
- Dominio V lega a: Eparina; Fibulina 2; Distroglicano α
- Dominio IV lega a: Entactina 1 e e 2; Fibulina 2;
Fibronectina; Eparina
- Localizza l'acetilcolinesterasi alle NMJ
- Protegge i componenti delle cartilagini dalla idrolisi
- Tipo 1A
- Insorgenza
- Infantile, Solitamente < 3 anni
- Difficoltà respiratorie e ad alimentarsi
- Prenatale
- Deglutizione danneggiata: Polidramniosi; Assenza della bolla dello stomaco
- Scheletro: Contratture; Femori corti
- Caratteristiche cliniche
- Neuromuscolare
- Rigidità muscolare
- Miotonia: Insorgenza dopo la nascita
- Faccia: Espressione fissa , Labbra aggrottate, Bocca piccola
- Cosce
- Camminata ondeggiante
- Posizione rannicchiata
- Trattamento: Carbamazepina
-
Ipertrofia muscolare: Specialmente cosce; Generalizzata
- Ipertermia maligna: Rischio aumentato
durante anestesia
- Riflessi tendinei: Ridotti
- Ritardo mentale: Lieve; Raro
- Scheletro
- Displasia ossea (moderata)
- Micrognazia
- Spina: Platispondilite; Emispondilo
- Altezza ridotta
- Cifoscoliosi
-
Contratture: Progrediscono fino alla media adolescenza
- Oculare
- Miopia
- Catarrate
- Blefarofimosi
- Dispiazzamento mediale del canthi esterno
- Irsutismo
- Testicoli piccoli
- Problemi per l'anestesia
- Difficoltà
meccaniche durante intubazione: Microstomia e rigidità dei muscoli
della mascella
- Limitazione delle escursioni respiratorie: Deformità della spina e gabbia toracica
- Laboratorio
- EMG: Continue scariche muscolari
- Miotonia o scariche ripetitive complesse
- Presenti dall'età di 7 mesi
- Non modulazione della frequenza o ampiezza
- ? Attività dovuta a basi muscolari o nervose
- CK serico: Lievemente alto o normale
- Biopsie muscolari
- Istochimica: Alterazioni dei tipi di fibre; Nuclei interni; Fibre di
dimensioni variate
- Perlecan: Ridotte o mutate molecole secrete dentro la matrice
extracellulare
- Raggi
X: Osteocondrodisplasia
- Tipo 1B
- Le mutazioni
Perlecan
- Insorgenza: Nascita
- Più grave displasia ossea: Simile alla Kneist
Sindrome Schwartz-Jampel Tipo 2/Stüve-Wiedemann (SWS)
26
l Recettore
del fattore inibitorio leucemico (LIFR)
; Cromosoma 5p13.1; Recessiva
- Genetica del
LIFR: Le mutazioni nulle
-
Proteina recettore LIF: Lega numerosi citochine
- LIF
- OSM
- Fattore neurotrofico ciliare (CNTF)
- Cardiotrofina 1
- Neurotrofina cellulare 1/B stimolante il fattore 3
- Insorgenza: Nascita
- Clinica
- Congenita
- Ipotonia
- Difficoltà respiratorie e di alimentazione
- Episodi di ipertermia
- Alta mortalità nell'infanzia
- Scheletro
- Contratture delle articolazioni
- Displasia ossea a legno verde: Campomelia metafisale (simile alla
malattia di Pyle
)
- Arti inferiori ad arco
- Inspessimento corticale interno
- Metafisi larghe: Modello trabecolare anormale
- Campodattilia
- Fratture spontanee
- Bassa statura
- Autonomo
- Ipertermia maligna: Episodi ricorrenti
- Instabilità della temperatura
- Riflessi cornei: Assenti
- Lingua liscia
- ? Patogenesi: ? Dovuta a danneggiamento delle segnalazioni di
STAT3
- Ridotta induzione delle proprietà colinergiche e peptidiche
dei neuroni simpatici
- Riflessi tendinei: Ridotti
- Laboratorio
- Topo knockout LIFR
- Riduzione del volume osseo fetale: Aumentato numero degli osteoclasti
- Riduzione del numero degli astrociti nel midollo spinale e encefalo
- Morte perinatale
Malattia di Brody
l
Ca++ ATPasi (ATP2A1)
;
Cromosoma 16p12; Recessiva
- Le mutazioni nel gene ATP2A1 5
- Presente in 6 di 8 famiglie con sindrome di Brody recessiva
- Tipi
- Stop o frameshift in 5 famiglie
- Missenso con ridotta funzione in 1 famiglia (Pro789Leu)
- Non mutazioni ATP2A1 in 2 famiglie dominanti
- Proteina Ca++ ATPasi
- Localizzazione: Reticolo sarcoplasmatico per la contrazione veloce
dei muscoli scheletrici
- Più basso Ca++ citoplasmatico: Pompa Ca++ nello
spazio luminale o extracellulare
- Promuove il rilassamento delle fibre muscolari dopo la contrazione
- Isoforme neonatale e adulta prodotte da splicing alternativo
- Distribuzione: Fibre muscolari di tipo 2
- Struttura: HPRD
- Epidemiologia
- Preponderanza maschi
- Rara: 1 su 10.000.000 di nascite
- Caratteristiche cliniche
- Insorgenza: Infantile; Muscoli
degli arti
- Rilassamento muscolare compromesso; Rigidità; Crampi e Mialgie
- Indotta da esercizio
- Insorgenza: Minuti dopo l'inizio dell' esercizio
- Peggiora con il progredire dell' esercizio
- Freddo: Sintomi peggiorano
- A riposo: Miglioramento dei sintomi dopo secondi o minuti
- Crampi
senza dolore
- Distribuzione: Arti e
Faccia (Palpebre)
- Forza
- A riposo: Normale
- Durante esercizio: Ridotta
- Dopo esercizio: Forza ritorna normale entro minuti
- Dimensioni muscolari: Possono essere ingrossati
- Decorso: Lenta progressione; Rara la
debolezza permanente
- Rabdomiolisi in alcuni pazienti
- Laboratorio
- CK serico: Può essere lievemente elevato
- EMG
- Crampi elettricamente silenti
- Unità motorie normali
- Patologia muscolare
- Atrofia di tipo II
- Non alterazioni miopatiche
- Assenza di colorazione ATP2A1 (SERCA 1) con mutazioni per troncazione, ma non
per missenso
- Trattamento: ? Dantrolene; Nifedipina; Verapamile
- Clinica variante: Sindrome di Brody
- Ereditarietà: Può essere dominante
- Insorgenza nell'adolescenza
- Coinvolgimento muscolare selettivo
- Non mutazioni ATP2A1
l
Ereditarietà dominante
- Epidemiologia: Famiglia
italiana
- Genetica: Cosegregata con una traslocazione costituzionale apparentemente bilanciata (2;7)(p11.2;p12.1)
Miochimia e
epilessia neonatale
benigna
13
l
KCNQ2
; Cromosoma 20q13.3; Dominante
- Epidemiologia: Una famiglia tedesca
- Mutazione: R207W
- Fisiologia: Mutazione
- Spinta dei voltaggio-dipendenti a voltaggi più positivi
- Rallenta l'attivazione di quelli indotti dalla
depolarizzazione
- Clinica
- Insorgenza: Convulsioni
neonatali
- Mialgie: Fanciullezza
- Miochimia
- Aumenta con età
- Esacerbata da: Febbre, freddo, alcol e gravidanza
- Distribuzione: Tronco; Arti; Non faccia
- EMG
- Scariche spontanee di gruppi di potenziali di unità
motorie
- Scariche ripetitive dopo stimolazione del nervo
- Attività inibita dal bloccaggio delle NMJ
- Trattamento: Carbamazepina
- Epilessia neonatale benigna
Mioedema
- Rigonfiamento localizzato, persistente, elettricamente silente dei muscoli dopo percussione
- Durata: Pochi secondi
- Diversa dalla increspatura dovuta a miotonia
- Dovuta alla contrazione locale dopo il rilascio del calcio dal reticolo sarcoplasmatico
- Malattia associate: Cachessia;
Mixedema; Iponatremia;
Rabbia; Occasionali normali
Spasmi muscolari e Dwarfismo
Spasmi muscolari
familiare e Dwarfismo
l
Recessiva; ? Autosomica o collegata al cromosoma X
- Caratteristiche cliniche
- Insorgenza: Infantile
- Contrazioni muscolari
dolorose di tronco e arti
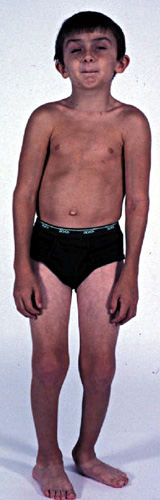
- Bassa statura, naso grosso e orecchie posizionate in basso
- Capelli: Scalpo - sottili; Corpo - nessuno
Sindrome di Satoyoshi
- Epidemiologia
- Frequenza: 30 casi descritti
- Sporadica: Tutti i pazienti
- Insorgenza: 6 a 15 anni
- Muscoli
- Spasmi muscolari: Progressivi, dolorosi, intermittenti
- Mioglobinuria
- Sistemico
- GI (50%): Diarrea; Malassorbimento
- Endocrino: Amenorrea; Utero e ovaie ipoplastici
- Alopecia universale
- Scheletro
- Patologia ossea: Distacco epifisario ; Cisti; Fratture da fatica; Osteolisi
- Bassa statura
- Trattamento: 1 paziente migliorato dopo corticosteroidi 23
Schwartz-Jampel
Crampi
- Caratteristiche cliniche
- Insorgenza acuta
- Breve durata
- Contrazione muscolare: Forte e palpabile
- Sollievo dallo stiramento dei muscoli
- Tipo comune: Nei normali
- Contrazione muscolare improvvisa, involontaria, dolorosa
- Precipitanti
- 2° a ipereccitabilità della arborizzazione
terminale del nervo
- Più spesso a seguito di contrazioni volontarie: Specialmente
contrazioni di accorciamento
- Sporadicamente durante riposo o sonno
- Stimolazione elettrica a
10 Hz
- Sollievo: Stiramento muscolare passivo; Massaggio locale
- Localizzazione
- Solitamente coinvolgono singoli muscoli più frequentemente che gruppi
- Specialmente gastrocnemio
- Crampi locali in altri muscoli spesso associati a denervazione o altre malattie neuromuscolari
- Sequela comune
- Muscoli indolenziti e gonfi
- CK serico alto
- EMG
- Potenziali di azione trifasici: Irregolari
- Frequenza: 40 a 60 Hz
- Origine: Terminali nervosi motori
- Malattie sistemiche associata con i crampi
- Malattie neuromuscolari associate con i crampi
- Malattie
dei crampi familiari
- Trattamenti per crampi
|
Normalizzare le anomalie metaboliche
Solfato di chinino:
260 mg qhs o bid
Carbamazepina:
200 mg bid o tid
Fentoina:
300 mg qd
Tocainide:
200 a 400 mg bid
Verapamile:
120 mg qd
|
Amitriptilina:
25 a 100 mg qhs
Vitamina E: 400 IU qd
Riboflavina: 100 mg qd
Difenildramina:
50 mg qd
Calcio: 0.5 a 1 g elementare Ca++ qd
|
- Vedi anche: Dolore muscolare
Attività elettrica con poche manifestazioni cliniche
Crampi
elettricamente silenti
Muscoli Attività: Origine neurale
Tetanus
20
Caratteristiche cliniche
7
- Epidemiologia
- Femmine (70%) > Maschi (30%)
- Prodromi: Dolori e rigidità nei muscoli
assiali
- Insorgenza
- Età
- Fanciullezza o adulti
- Positiva agli anticorpi anti-GAD: Media 41 anni. Più
comune 3a a 5a
decade
- Prodromi: Episodica rigidità o cadute
- Decorso: Lentamente progressivo (insidioso); Talvolta insorgenza rapida
- Riportate forme ereditarie
:
Può essere hyperekplexia
- Rigidità
- Distribuzione
- Specialmente nei muscoli assiali e prossimali
degli arti
- Asimmetrica, o preminente in 1 gamba (30% a 75%)
- Coinvolgimento degli arti compromette il camminare
- Iperlordosi: Lombare; Limita la flessione del tronco
- Fluttuante nel tempo: Ridotta nel sonno
- Spasmi muscolari
- Scatenati
- Stiramento
- Emozione
- Stimolazioni sensorie
- Paura degli spazi aperti
- Può avvenire spontaneamente
- Decorso dello spasmo
- Scatto mioclonico improvviso: Può provocare cadute
- Seguito da attività toniche che sussistono per secondi
- Solitamente bilaterale
- Movimenti tipici
- Braccia: Estensione e pronazione
- Tronco: Estensione
- Gambe: Estensione e lieve abduzione; Inversione dei piedi
- Co-contrazione di agonisti e antagonisti
- Muscoli paraspinosi addominali e toracici
- Spasmi possono produrre piccoli movimenti dovuti alla co-contrazione attorno
alle giunture
- Sofferenza: Può essere associato con grave dolore
- Possono essere gravi abbastanza da causare fratture
- Rilassamento degli spasmi: Sonno e benzodiazepine
- Disabilità: Bastone o deambulatore comunemente necessari 2°
alle frequenti cadute
- Riflessi
- Riflessi tendinei: Normali o aumentati
- Cutanei addominali: Possono essere persi
- Risposte Startle: Aumentate
- Andatura: "Soldatino di stagno"
- Disfunzione autonoma
- Movimenti extraoculari: Alcuni pazienti
- Nistagmo con
sguardo conservato
- Abduzione limitata
- Disallineamento oculare verticale e orizzontale
- Carenza dei movimenti lisci
- Iniziazione saccadica danneggiata
- Esame neurologico altrimenti normale: La maggior parte dei pazienti
- Decorso della malattia
- Solitamente: Lenta progressione o statico per anni
- Sindromi focali possono progredire
ad un coinvolgimento più generalizzato
- Morte
improvvisa: Alcuni pazienti
-
Trismo insolito
- Aritmie cardiache
- Arresto
respiratorio restrittivo
- Rapida riduzione del baclofene intratecale
Malattie associate
Laboratorio
- CK serico: Transientemente elevato
- ANA (30%)
- CSF
- Bande oligoclonali (60%)
- Proteine alte (20%)
- Pleocitosi: 10% nella SPS; 63% nella variante PERM
- Normale: 40% nella SPS; 12% nella variante PERM
- Anticorpi anti-GAD: Possono essere presenti
- Solitamente a titolo più basso che nel siero
- Può aversi sintesi intratecale anti-GAD
- MRI CNS: Non-diagnostica
-
EMG
- Continui potenziali di azione
- Indistinguibili dalla attività volontaria: Unità motorie normali
- Persiste durante i tentativi di rilassamento
- Maggiormente preminenti nei muscoli assiali
- Treni d'onda persistenti: 50 a 60 ms durata; 5 - 6 Hz; Ritmici; Sincroni
- Interruzione dei treni d'onda negli spasmi: Attività rapida; Modello della piena interferenza; > 4 sec
- Attività ridotta da diazepam intravenoso
- Non fibrillazioni o scariche ritmiche raggruppate
- Scarso rilassamento dopo contrazione
- Patologia del CNS
- Infiammazione perivascolare
- Midollo spinale: Perdita neuronale
- Nucleo vestibolare laterale: Perdita di neuroni
Meccanismi della malattia
- Patofisiologia
- Malattia della inibizione dei terminali presinaptici Ia
- Riflessi polisinaptici esagerati
- ? Difetto nel controllo dei percorsi discendenti
- Normale eccitabilità dei neuroni motori el a funzione delle
cellule di Renshaw
- Diversa dal tetano il quale produce
una inibizione segmentale difettosa e ipereccitabilità dei neuroni motori
- Farmacologia e percorsi
- Percorsi
discendenti monoaminergici reticolospinali
- Inibizione dei riflessi flessori spinali afferenti
a latenza breve
- Percorsi dei flessori spinali a latenza lunga
facilitati
- ? Relativa sovraattività nei SPS
- Inibizione monoaminergica riduce spasmi e rigidità: Clonidina; Tizanidina
- Attività GABA: Inibitoria
- ? Relative sottoattività
- Inibizione GABA-ergica aumentata riduce spasmi e rigidità: Baclofene; Diazepam
Anticorpi
- Decarbossilasi acido glutammica (GAD)
- Spettro di età: Adulti
- Modello di colorazione tissutale:
Terminali nervosi GABAnergici
- Titolo anticorpale anti-GAD
- Livelli: Basso ad alto
- Varia con le sindromi cliniche
- Alto nella sindrome di dell'uomo rigido (Stiffman)
- Localizzazione degli anticorpi
- Presenti nel siero e CSF
- Solitamente titolo più alto nel siero
- Struttura degli epitopi GAD: Lineare e conformazionale
- Bersagli GAD: Varianti 65 e 67 kD
- Sindrome di dell'uomo rigido (Stiffman)
- Prevalenza: 50% a 90%
- Specificità: Maggiore con titolo più alto di anticorpi
anti-GAD
- Altre sindromi: Anticorpi anti-GAD trovati anche
- Miocloni
palatali
- Epilessia: Resistente alla terapia, correlate
alla localizzazione
- Frequenza: 15%
- Titolo degli anticorpi anti-GAD : Alto o moderato
- Anticorpi anti-GAD non trovati nella epilessia
generalizzata
- Atassia cerebellare:
Può avere un titolo più alto di anticorpi anti-GAD
-
Diabete mellito insulino-dipendente (IDDM)
- Spettro di età: Fanciullezza e adolescenza
- Titolo degli anticorpi anti-GAD: Titolo da basso
a moderato
- Struttura degli epitopi GAD: Conformazionale
- Bersagli GAD: Varianti 65 kD
- Associazione con
- Anticorpi alla tirosina fosfatasi IA-2
- Cellule immunitarie T
- Sindrome autoimmunitaria poliendocrina II
- Altri anticorpi nella sindrome di dell'uomo rigido (Stiffman)
- Cellule delle isole pancreatitiche (60%)
- Trovati anche nel diabete di tipo I: Titolo più basso, differente modello di colorazione
-
Anfifisina (128kDa)
:
Associata con il cancro della mammella
- Proteina 80 KDa
-
Citoplasma delle cellule Purkinie
- Bande oligoclonali nel CSF: 50%
- Collegamento esterno:
Yale
Terapia (Raramente completamente efficace)
- Sintomatica
- Diazepam (dosi molto alte; 20 mg a 300 mg/giorno); Clonazepam
- Può esacerbare la depressione
- Baclofene
- Baclofene (orale): Aggiunta di farmaci
- Pompa del Baclofene (intratecale)
- Indicazioni: Gravi sindromi
- Svezzamento improvviso può essere fatale
- Acido valproico
- Tiagabine: 6 mg qd
- Terapie immuno-modulanti
- Corticosteroidi: Alte dose solumedrol con riduzione; Miglioramento
su mesi
- Immunoglobuline
intravenose
- Cambio
del plasma
- Rituximab31
Sindrome della persona rigida: Varianti
- SPS focale: Solitamente sindrome
delle gambe rigide
- Insorgenza: 35 a 60 anni
- Rigidità e spasmi degli arti inferiori
- Stimoli: Specialmente movimenti volontari; Anche stimoli
riflessi
- Asimmetrica
- Postura dei piedi
- Tronco risparmiato
- Anticorpi anti-GAD: Presenti
- Trattamento (Parziale): Baclofene e diazepam
- Sindrome delle braccia rigide : Può suggerire malattie
paraneoplastiche
- Encefalomielite subacuta (tronco cerebrale) (PERM)
- Insorgenza: Subacuta (settimane)
- Clinica
- Rigidità
- Distale > prossimale all'insorgenza della malattia
- Rigidità
- Spasmi: Episodici
- Deperimento muscolare e debolezza
- Autonomo
- Iperidrosi associata con spasmi
- Può aversi insufficienza
- Nervi craniali
- EOM: Nistagmo e oftalmoplegia
- Cecità
- Sordità
- Disartria
- Altro CNS
- Tratto lungo
- Vertigini
- Intelletto preservato
- Progressione
- Mesi
- Morte in 1 a 39 mesi
- Alcuni casi lievi
- Alcuni casi possono evolversi dalla presentazione SPS
- Laboratorio
- CSF: Infiammatorio
- Neurofisiologia: Simile alla SPS; Miocloni
tronco cerebrali
- Patologia
- Infiammazioni: Perivascolari
- Gliosi nel midollo spinale, ponte e medulla
- Perdita neuronale: Grigia spinale
- Sindrome
Jerking della persona rigida
- Clinica
- Rigidità: Assiale e arti inferiori
- Caratteristiche successive
- Miocloni
tronco cerebrali
- Segni dei motoneuroni superiori
- PERM
- Progressione su decadi
- SPS paraneoplastico: Caratteristiche
indicative
- SPS confinata agli arti superiori
- Malattia con progressione rapida: A deformità fissata delle
articolazioni
- Associata a gangliopatia sensoria
- Naoplasmie: Mammella; Cellule polmonari piccole
- Anticorpi:
Anfifisina I; GAD
- Sindrome cerebellare con anticorpi anti-GAD 27
- Epidemiologia
- Insorgenza età: 20 a 74 anni
- Femmine
- Clinica
- Insorgenza: Lentamente progressiva
- Atassia: Arti e Tronco; Nistagmo; Disartria (50%)
- Rigidità (15%)
- Non coinvolgimento del tronco cerebrale
- Malattie associate
- Diabete mellito ID (a tarda insorgenza)
- Tiroidite
- Sindrome
poli endocrina
- Trattamento: Non descritto
- Laboratorio
- Anticorpi anti-GAD: Titolo alto
- Cellule Ab anti-parietali : Comuni
- MRI: Non-diagnostica
- Vedi anche:
- Iperekflessia (sindrome del bambino rigido)
- Insorgenza nella fanciullezza: mutazioni gene DYT1
- Escludere: Anemia perniciosa associata
Stricnina
11
- Azioni
- Blocca i recettori della glicina: Antagonista competitivo
- Blocca i neuroni inibitori nel tronco cerebrale e midollo spinale
- Fonti
- Topicidi
- Adulterazione da spacciatori di droga
- Trattamenti erboristici: Fatti con Strychnos nux-vomica
- Metabolismo
- Tipo chimico: Alcaloide
- Assunzione: Orale con assorbimento nel GI
- Metabolismo epatico: Emivita 10 a 16 ore
- Qualche secrezione urinaria: Fino al 20%
- Sindrome clinica
- Insorgenza: Acuta; 10 a 20 minuti dopo l'ingestione
- Segni iniziali: Nervosismo; Iperattenzione; Confusione
- Rigidità
- Inizio:
Faccia e collo
- Successivamente: Rigidità generalizzata
- Può essere scatenata dal minimo stimolo
- Durata: 1/2 a 2 minuti
- Durante lo spasmo può aversi arresto respiratorio
- Contrazione muscolare può causare rabdomiolisi
- Riflessi tendinei: Aumentati
- CNS: Agitazione; Delusioni; Disorientamento
- Diagnosi: Tossicologia urinaria (cromatografia urinaria)
- Diagnosi differenziale: Tetano (Non CNS)
- Trattamento
- Lavaggio gastrico
- Rigidità: Benzodiazepine; Barbiturici
- Rabdomiolisi: Fluidi; Gestione del pH
- Ventilazione profilattica nei casi gravi
- Vedi anche: Iperekflessia ( Malattia Startle)
- Collegamenti esterni:
USDA
Iperekplexia (
Malattia Startle)
l
Recettore della glicina,
subunità α-1 (legante strichinina)
; Cromosoma 5q33-q35;
Dominante o recessiva
- Caratteristiche cliniche
- Rigidità e miocloni dopo stimolo improvviso
- Insorgenza:
Già nell'infanzia con ipertonia o miocloni
- Insorgenza e gravità varie all'interno delle famiglie
- Trattamento: Clonazepam
- Modello di topo: Spasmodic e Oscillator
Fibrillazioni
- Potenziali di azione spontanei in una singola fibra muscolare
- Non visibile all'esame fisico
- Dimensione: 1 a 5 msec durata; 20 a 200 μV ampiezza
- Forma d'onda: Difasica o trifasica con iniziale positività
- Differisce dai potenziali di placca motrice che hanno iniziale negatività
- Velocità di emissione: 1 a 30 al secondo; Media 13; Solitamente regolare
- Aumenta con riscaldamento muscolare e inibitori
della colinesterasi
- Causa
- Comune: Diminuito potenziale di riposo di membrana nei muscoli
denervati
- Altro: Miopatia infiammatoria; Neonati
Fascicolazioni
- Scariche spontanee in un assone causante contrazione delle fibre muscolari in unità increspate
- Produce visibile increspature dei muscoli
- Possono originare in qualsiasi punto lungo il corso degli assoni
- Causa
- Benigna
- Comune nel gastrocnemio in pazienti altrimenti normali
- Forza normale nei muscoli in fascicolazione
- Esacerbata da esercizio, iperventilazione e tensione
- Può essere associata con la tendenza ai crampi
- Neuropatie motorie e malattie dei neuroni motori
- Malattie metaboliche:
Iperparatiroidismo;
Ipertiroidea; Ipomagnesemia
- Farmacologica
- Anti-Colinesterasi;
Caffeina;
Teofillina;
Terbutalina;
Litio
- Minipolimiocloni: Movimenti di piccola ampiezza,
specialmente delle dita di mani e piedi, dovuta a fascicolazioni di unità motorie
ingrossate
- Treni d'onda di potenziali di azione muscolari complessi emessi
ricorrentemente
- Scariche di gruppi di fibre muscolari
- Attivate efapticalmente
- Emissione in un modello regolare
- Inizianti spontaneamente o a seguito di irritazione
muscolare
- Cadenza
- Gamma: 5 a 100 Hz
- Frequenze più comuni: 30 a 40 Hz
- Più consistenti durante i treni d'onda che nella miotonia
- Interruzione dell'emissione e fine
- Treni d'onda non-ritmici
- Spesso associati con radiculopatia e malattie croniche
- Raramente produce ipertrofia muscolare (solitamente unilaterale)
Tetania con spasmo carpopedale
- Patofisiologia
- Anormale controllo del Ca++ dai canali
Na+
- Ipereccitabilità assonale: Dovuta a
- Depolarizzazione delle membrane
- Ridotta soglia dei potenziali di azione
- Scariche spontanee ripetitive
- Frequenza: 5 a 15 Hz le più comuni
- Fonte: Sorgono negli assoni periferici
- Causa
- Alterazioni
ioniche
- Alcalosi: Respiratoria
- Tetania normopotassica: Rara
- Acquisita: Associata con neoplasmia
- Ereditaria: Dominante con epilessia
- Esacerbata da: Iperventilazione; Ischemia
-
EMG
- Scariche assonali spontanee ad alta cadenza: Fino a 300 Hz
- Scariche ripetitive: Doppiette o multiple
- Spasmo intenso: Modello
di
interferenza
massimale
- Normali doppiette avvengono a < 10 Hz
- Diagnosi: Esame dell'avanbraccio ischemico
- Trattamento: Calcio; Magnesio; Fentoina
|
Barker |
Geniospasmo
l
Cromosoma 9q13-q21; Dominante
- Movimenti verticali della punta del mento e tremolanti del labbro inferiore
- Avvengono spontaneamente; O con stress, concentrazione, emozione
- Durata: Minuti
- Insorgenza: Infanzia e prima fanciullezza
- Più comune: Fanciullezza e adolescenza
- Associata col morso notturno della lingua
- Ridotta frequenza con l'aumento dell'età
- Altre causa del mento tremolante:
BSMA; Sindrome Orale-facciale-digitale I
-
Sindrome
dello spasmo palmaris brevis
2
|
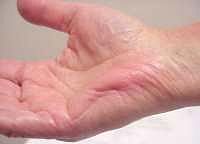
dal: M. Al-Lozi
|
|
Spasmo palmare breve |
- Muscoli
palmari brevi
- Localizzazione: Ipo-eminenza tenar; Sottocutaneo
- Nervi: Ulnare, Branca superficiale
- In azione: Rilievi e increspature nella cute soprastante
- Non sotto controllo
volontario
- Caratteristiche cliniche
- Epidemiologia
- Età di insorgenza: 37 a 76 anni
- Maschi > femmine
- Scatenata: Danneggiamento o intrappolamento dei nervi; Idiopatica
- Contrazioni dei muscoli palmaris brevis
- Localizzazione dell'attività:
- Isolata ai muscoli palmaris brevis
- Bilaterale o unilaterale
- Nessuna negli altri muscoli
- Spontanee
- Tempi: Irregolari
- Produce increspatura del lato ulnare della mano
- Influenze
- Aumentano le contrazioni: Stress; Eccitamento; Sporadicamente muovendo
il 5o dito
- Diminuiscono le contrazioni: A riposo; Nel sonno; Stiramento
dei muscoli; Blocco del nervo distale
- Alterazioni sensorie: Variabili
- Sistemico (Alcuni pazienti): Mialgia e intolleranza all'esercizio
- Decorso: Benigno; Nel corso di anni
- Trattamento: Dilantina in 1 paziente
-
EMG
- A riposo: Scariche ad alta frequenza con irregolarità
- Unità motorie: Normale ampiezza, durata e morfologia
-
Miochimia
dell'obliquo superiore
- Malattia della motilità oculare
- Sintomo: Oscillopsia parossistica
- Movimenti torsionali e verticali monoculari
- Lateralità predominante a destra: 80%
- Dovuta a contrazioni intermittenti del muscolo superiore
obliquo
- Decorso: Ricorrente
- EMG: Treni d'onda spontanei dei potenziali della unità motorie
- Patologia: Contatto arteriale alla radice della zona di uscita
del sintomatico
nervo
trochleare (IV) 19
Distonia rispondente
alla
Dopa
Sindrome di Crisponi
l
Fattore 1 simile al recettore citochina (CRLF1)
; Cromosoma 19p12;
Recessiva
- Genetica: Le mutazioni CRLF1 anche nella
sindrome della sudorazione fredda indotta
- Insorgenza congenita
- Clinica
- Movimenti: Spesso episodici
- Contrazione dei muscoli facciali: "Trisma"
- Stimoli: Stimolo tattile e pianto
- Ipertonia dei muscoli del collo e opistotono
- Contrazioni muscolari generalizzate: Episodi
- Avvengono numerose volte al giorno
- Stimoli: Esterni
- Attacchi epilettici
- Ipertonia
- Anomalie
- Faccia: Faccia grande, guance paffute, naso largo
- Campodattilia
bilaterale
- Scoliosi
- Disturbi respiratori: Apnea centrale; Ipopnea
- Febbre: Episodica
- Difficoltà di alimentazione
- Decorso
- Eccitabilità può diminuire oltre il 1o anno
- Morte: Alcuni pazienti; < 1 mese alla fanciullezza
Whipple
- Scatti mioclonici oculari e facciali (mioritmia)
- Oftalmoplegia: Sopranucleare
- Demenza
Sindrome
dei crampi muscolari: Autosomica Dominante
Crampi muscolari e sindrome neuropatica 1
l
Autosomica Dominante
- Crampi
- Insorgenza: Fanciullezza ai 20 anni
- Picco dei sintomi: Adolescenza e 20 anni
- Durata: 2 a 3 minuti
- Dolorosi; Arti e tronco
- Induzione
- Movimenti ripetuti o mantenere una postura specifica
- Avvengono anche durante il pieno rilassamento e sonno
- Elettrofisiologia
- EMG: Denervazione cronica; Non fascicolazioni o miochimia
- NCV: Lieve riduzione delle ampiezze di SNAP e CMAP nelle gambe
- Biopsia dei nervi: Ridotto numero degli assoni mielinizzati
Sindrome del crampo familiare
l
Autosomica Dominante
- Insorgenza età: 10 a 15 anni
- Crampi scompaiono dopo 25
anni di età
- CK serico: Alto
- Patofisiologia: Origine neurogena
- Biopsie muscolari: Denervazione
Vedi anche
Sindromi
dei crampi sporadici
Sindromi fascicolazioni-crampi
- Insorgenza: Giovani fino alla media età adulta
- Clinica
- Polpacci e quadricipiti
- Associata a dolore muscolare e fascicolazioni
- Forza normale
- Laboratorio
Crampi associati alla
gravidanza
- Frequenza: 10% a 30% delle gravidanze
- Insorgenza: Più comuni nel 2o trimestre
- Sintomi
- Durata: ~ 8 settimane
- Frequenza: Un giorno si ed uno no
- Localizzazione: Gambe
- Più comune
- Con il progredire della gravidanza
- Alla notte: Avviene raramente, solo durante il giorno
- ? Associazione con basso livello del magnesio serico
- Trattamento
- ? Lattato o citrato di magnesio presi al dosaggio di 5mmol
al mattino e 10mmol alla sera
- Non correlata ad
- Altre complicazioni di gravidanza
- Esito fetale
Ritorno a
Neuromuscolare home page
Ritorno a
Ion
Canale Malattie
Ritorno a
Indice delle miopatie e delle giunzioni neuromuscolari
Riferimenti
1.
J Neurol Neurosurg Psychiatry 1999;67:116-119
2.
J Neurol Neurosurg Psychiatry 1995:59:182-184
3.
Neuromuscular Disorders 1999;9:604-607
4.
Neuromuscular Disorders 1997;7:241-249
5.
umana Genetics May 2000
6.
urology 2000;55:383-388
7. Neurology 2000;55:1531-1555,
Movement Disord 2002;17:853-866
8.
J Neurol Neurosurg Psychiatry 2000;69:110-113
9.
Neurology 1999;52:1453-1459;
Muscle Nerve 2001;24:340-344
10.
Ann Neurol 2001;49:486-492
11.
NEJM 2001;344:1232-1239
12.
Science 2001;293:864-867
13.
PNAS 2001;October On-Line
14.
Muscle Nerve 2001;24:1542-1547
15.
Neurology 2001;57:1849-1857
16.
Muscle Nerve 2002;S11:S55–S58,
Brain 2002;125:1887-1895
17.
Muscle Nerve 2002;S11:S103–S107
18.
Brain 2002;125:2392=2407
19.
Ann Neurol 2002;51:361–368
20.
Neurol India 2002;50:398-407
21.
Ann Neurol 2003; Online March
22.
Muscle Nerve 2003;27: 449–455
23.
Am J Med Genet 2003;118A:52–54
24.
BMC Neurology, Malattie neuromuscolari 2002;13:347–351
25.
Muscle Nerve 2002;26:702-707
26. Am J Hum Genet 2004; February
27.
Arch Neurol 2001;58:225-230
28. Cervello 2004; Online July
29.
Arch Neurol 2004;61:1938-1942
30.
Muscle Nerve 2005; Online February
31.
J Neurol Neurosurg Psychiatry. 2005;76:999-1001
32. J Neurol 2006; Online March
33.
Infection 2006;34:35-38
17 ottobre 2006
Va a Acronimi e sigle
Va a Mitolario in
italiano
Va a Mitolario italiano -
inglese
Va a Mitolario inglese -
italiano