 Traduzione di Natale Marzari
Traduzione di Natale Marzari
Home,
grave,
Indice
alfabetico,
Collegamenti,
Patologia,
Molecole,
Sindromi,
Muscoli,
Giunzioni neuromuscolari,
Nervi,
Spinale,
Atassia,
Anticorpi e Biopsia,
Informazioni per i
pazienti
giovedì 29 aprile 2021 |
 |
fanciullezza insorgenza POLYNEUROPATHIES
Vedi accumulo inclusioni
|
STORAGE INCLUSIONS |
|---|
|
Tipo |
Malattia |
Localizzazione cellulare |
|
Osmophilic
con periodicity

Bilbao
|
Leucodistrofia metacromatica |
Cellule di Schwann
Macrofagi
Assoni |
|
Di Fabry |
Vascolare
Perineurial
Cellule di Schwann, non-mielinizzanti |
|
Niemann-Pick, Tipo 1 |
Tutti eccetto assoni |
|
Batten-Kufs |
Cellule di Schwann
Vascolare
Fibroblasti |
Tossici (lisosomiali)
Amiodarone
Clorochina
Peressilina
Simili inclusioni: Cockayne
|
Vascolare: endoteliali e muscolatura liscia
Nervi: Perineurial;
Cellule di Schwann (Non-mielinizzanti) |
|
Corpi zebrati |
Leucodistrofia metacromatica |
Cellule di Schwann
Macrofagi |
|
Mucopolysaccharidosis |
Cellule di Schwann-mielinizzanti
Fibroblasti |
|
Di Fabry |
Vascolare
Perineurial |
|
Niemann-Pick |
Cellule di Schwann
endoteliali
Macrofagi |
|
Pi granuli |
Cellule di Schwann |
Membrane-legato
cleft o polygonal spazio
|
Krabbe |
Cellule di Schwann
Macrofagi |
|
Adrenoleucodistrofia |
Cellule di Schwann
Macrofagi |
|
Farber's malattia |
Cellule endoteliali |
|
Empty vacuoli |
La malattia di Tangier |
Cellule di Schwann, non-mielinizzanti |
|
GM1 gangliosidosi |
Vascolare
Fibroblasti |
|
I-cellule malattia |
Cellule di Schwann, non-mielinizzanti
Fibroblasti
Perineurial |
|
Mucopolysaccharidosis |
Fibroblasti
Vascolare
Cellule di Schwann |
|
Xantomatosi cerebrotendinea |
Cellule di Schwann |
|
Wolman's malattia |
Tutti |
|
Sialidosi, tipo 1 |
Cellule di Schwann |
Cockayne's sindrome

l
Cromosoma 5; Recessiva
-
Scheletro: Dwarfism; Microcefalia; lunga arti
-
Cute: Senile apparenza; Pigmentazione; Luce sensitity
-
Occhi: Atrofia ottica; Degenerazione retinica
; "Hollow" apparenza
-
Vascolare: Aterosclerosi; Ipertensione
-
CNS: Perdita di udito, Ritardo mentale, Idrocefalo e Disturbi dell'andatura
-
PNS
-
Riflessi tendinei: Ridotti
-
Deperimento muscolare
-
Patologia
-
ipomielinizzante neuropatia
-
Inclusioni: Cellule di Schwann; lisosomiali, membrane-legato; Vacuoli con materiale granulare
Farber's malattia (lipogranulomatosi)
l
Acid ceramidase
 ;
Cromosoma 8p22-p21.3;
Autosomica recessiva
;
Cromosoma 8p22-p21.3;
Autosomica recessiva
-
Clinica
-
Nervi: Ipotonia; Distale atrofia
-
Cute: lipogranulomatosi; Noduli: periarticolare subcutaneous
-
CNS: Mental e ritardo motorio; Hoarse pianto
-
Sistemico: Epatomegalia; Splenomegalia; Nefopatia
-
Articolazioni: Doloranti, Rigonfi
-
Occhi: Macular cherry-rosso spots
-
Prognosi: Insufficienza respiratoria; Morte 2 a 20 anni
-
Elettrofisiologia
-
Velocità di conduzione nervosa: Lenta
-
Laboratorio
-
Neuronali e gliale accumulo di nonsulfonated acido mucopolisaccaridi
-
elevato urine ceramide livelli
-
N-laurylsphingosine deacilase Carenza
-
Ceramidase Carenza
-
Pathology10
-
Sistemico: Granulomatoso infiltrazione di fegato, milza, polmones, e timo
-
Farber corpi: Curved, tubulare; Vacuolare e citoplasmatiche; Ceramide
-
Nervi
-
Ipomielinazione
-
Cellule di Schwann: Inclusioni
-
mielinizzanti: elettroni-lucent membrane-legato clefts
-
Non-mielinizzanti: Membrane legato, allungati corpi
Neuropatia con assoni giganti
l
Gigaxonin
;
Cromosoma 16q24; Recessiva
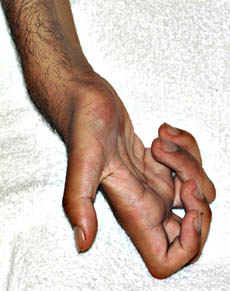
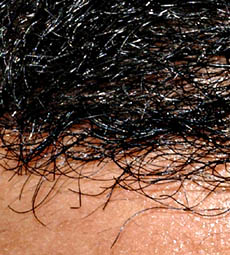
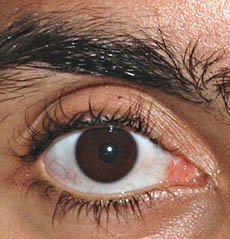
dal: T Mozaffar
Neuropatia con assoni giganti
|
-
Mutazioni genetiche; Missenso; Nonsensi; Frameshift
-
Gigaxonin proteina
-
Localizzazione subcellulare: Cytoskeletal
-
Legami alla microtubuli-associata proteina (MAP)1B catena sottile
-
Associata con microtubuli: Stabilizza microtubule network
-
Tessuto localizzazione: Espressione ubiquitaria
-
BTB/kelch superfamiglia
-
Intracellulare proteine
-
Funzioni
-
Coordinate cellule morfologia e crescita: Calicin
-
Citoplasmica sequestration di fattori di trascrizione: Keap 1
-
Associazione con Actina e citoscheletro
-
Può contribuire to virale patogenesi
-
Insorgenza
-
Età: Tipicamente 3 anni; La maggior parte di < 10 anni
-
Disturbi dell'andatura
-
Caratteristiche cliniche
-
Polineuropatia
-
Distribuzione: Gambe > Braccia
-
Debolezza: Diffusa; Distale > prossimale
-
Perdita sensoria: Specialmente vibratorie e proprioception
-
Riflessi tendinei: Ridotti in arti inferiori o Generalmente
-
Nervi craniali: Debolezza facciale; Disartria
-
Insorgenza
-
CNS: Variabile
-
Atassia: Clumsy andatura; Presente nella maggior parte pazienti
-
Occhi: Atrofia ottica; Nistagmo
-
Ritardo mentale
-
Tratto corticospinale: Risposta dell'estensore plantare
-
Capelli
-
Frizzy (tightly curled) scalp capelli; lunga curly eyelashes
-
Cambiamenti più comune in grave casi
-
Scheletro: Bassa statura; Preminente alto foreCapo
-
Progressione
-
Wheel chair o Morte by 3a decade
-
Alcuni pazienti con progressione più lenta over 20 anni
-
Laboratorio
-
CSF: Normale
-
MRI
-
Diffuse iperintensità: Cerebrale e cerebellare materia bianca
-
Elettrofisiologia: Neuropatia assonale
-
SNAPs: Assenti
-
Motorio velocità di conduzione nervosa: Normale o Lieve riduzione
-
Patologia
-
Perdita assonale: Distale; Progressiva con età; Tutti assoni dimensioni
-
Rigonfiamenti assonali
-
Contiene densely packed, swirled neurofilamenti
-
Inizio a nodi di Ranvier
-
fogli mielinici: Sottile o assente Attorno swellings
-
Dimensioni: Media 20 μM; Fino ai 50 μM
-
microtubuli distribuzione: ANORMALEely clustered
-
Assonale atrofia: Distale to swellings
-
Microfilament accumulazioni
-
In Cellule di Schwann, fibroblasti, vascolare endothelium
-
Eterozigoti
-
? Subclinical polineuropatia on elettrofisiologia
-
Varianti: Neuropatia assonale gigante, Tunisian Forma
l
Autosomica recessiva
-
Insorgenza: Infanzia
-
Clinica
-
Debolezza: Distale; Bulbare
-
Perdita sensoria: Gambe
-
Riflessi tendinei: Vivaci
-
Normale capelli
-
multisistema degenerazione con neuroni motori sindrome
-
Lenta progressione
-
Altro citoscheletrica Malattie
Distrofia neuroassonale (Tarda infantile)
l
Autosomica recessiva
-
Caratteristiche cliniche: Tipica sindrome
-
Insorgenza
-
1 a 2 anni
-
Andatura difficoltà
-
CNS
-
Perdita di mentale e motor pietra miliare
-
Attacchi epilettici
-
Aumentato tono
-
Extrapiramidale: Distonia; Rigidità
-
Spasticità: Alcuni pazienti
-
Atassia
-
Sordità
-
Neuropatia: Poorly definiti alla EMG o Patologia
-
arti mutilation
-
Ipotonia
-
Riflessi tendinei: Assenti o ridotta
-
Miopatia
-
No cliniche segni
-
CK Serico: lievemente elevata in alcuni casi
-
Patologia muscolare
-
Necrosi
-
Fibre splitting
-
Endomisiale macrofaga attivazione
-
Rigonfiamenti assonali in intramuscolari nervi e terminali
-
Endocrine: Il diabete insipido; ipotalamica hypothyroid
-
Autonomo
-
Perdita di tears (cheratite)
-
Scarsa regolazione della temperatura
-
Ritenzione urinaria
-
Decorso: Morte < 10 anni
-
Diagnosi: Gonfiori può essere presente on conjuctival, cute o Biopsia muscolare
-
Patologia
-
Spheroids: In CNS e Nervi periferici
-
Contenente: Membrane e anormale mitocondri
-
Colorazione: PAS positive
-
Nervi periferici
-
Gonfiori può essere presente
-
Sottili perdita assonale o Degenerazione walleriana
-
Ultrastrutture: Tubulovesicular profili; mitocondriale aggregati
-
CNS: Ferro deposito
-
Infantile variant8
-
Clinica
-
Movimenti: Hyperkinetic; Toniche spasmi
-
Attacchi epilettici
-
Parlata apneica
-
Decorso: Progressiva to ipotonia o plateau a 3 anni
-
Laboratorio
-
Cute nervi: Thnella mielina; condensati assonali filamenti
-
Cervello: Assoni terminale degenerazione in corteccia, gangli basali e cervelletto
Congenita ipomielinizzante Neuropatie
Neuropatia ipomielinizzante congenita
l
P0 proteina
;
Cromosoma 1q22; Recessiva
-
Clinica
-
Gravi generalizzato debolezza e degenerazione
-
Artrogriposi
-
Insorgenza: Nascita
-
Decorso
-
Casi gravi: Morte prima 3 mesi
-
più lievi casi: Staticoa debolezza; Aumentato disabilità con crescita
-
Elettrodiagnostica
-
Demielinizzanti: NVC molto lenta; Non blocco di conduzione
-
Da simile a HMSN III
-
Patologia: Sottile o assente fogli mielinici
Attorno assoni
Neuropatia ipomielinizzante congenita
l
CMT4E;
Precoce crescita risposta-2
;
Cromosoma 10q21.1-q22.1; Dominante o recessiva
-
Gene
-
Mutazioni
-
Ile268Asn (Omozigote); Ser382 Arg e Asp383Tyr (eterozigosi)
-
Recessiva mutazioni in repressor legante dominio (R1): ? alter trascriveional attività
-
Anche difettose in: Krox20 knockout topo;
CMT1; Dejerine-Sottas
-
Proteina: a dita di zinco
-
Clinica
-
Insorgenza: Infantile ipotonia
-
Debolezza: Diffusa; Distale > prossimale; Deambulazione ± walker
-
Degenerazione: Diffuse
-
Velocità di conduzione nervosa: 3 a 8 M/s
-
Patologia dei nervi
-
Mielina: Assenti o grandemente ridotta on maggior parte assoni
-
Bulbi a cipolla: Occasionali
-
Perdita assonale: Da lieve a moderata
Neuropatia ipomielinizzante congenita, Asintomatica
l
Rho Guanine-Nucleotide Excambiamenti Fattore 10 (ARHGEF10)
;
Cromosoma 8p23; Dominant14
-
Epidemiologia: Singola famiglia
-
Genetica: Mutazione missenso; Thr109Ile
-
ARHGEF10 proteina
-
Rho famiglia di GTPasi proteine (RhoGTPases)
-
Regola Actina citoscheletro
-
Influenza cellule sviluppo
-
Polarity; microtubuli dinamiche; Membrane-Percorsi di trasporto; trascriveion-fattore attività
-
Play ruolo in neuronale morphogenesis
-
Cell migrazione; Assonale crescita e guidance; Dendrite elaboration e plasticity; Synapse Formazione
-
Guanine-nucleotide excambiamenti fattore (GEF): Altre malattie
-
FGD1 GEF: Faciogenital displasia
-
ARHGEF6 (aPIX): Collegate al cromosoma X non sindromica ritardo mentale
-
ALS2 (alsin): autosomica recessiva sclerosi laterale amiotrofica
-
Clinica
-
La maggior parte di pazienti asintomatico
-
Esaminazione: Spesso normale: Normale riflessi tendinei e Massa muscolare
-
Pazienti occasionali con Raynaud's fenomeno
-
Esame elettrodiagnostico
-
Velocità di conduzione nervosa: Uniformi rallentamento; Braccia 33 a 42 m/s; Gambe 27 a 36 m/s
-
CMAPs: Normale ampiezza
-
SNAPs: Normale o lievemente ridotti in ampiezza
-
Simili valori a tutti all'età di: ? Non-progressivo
-
Patologia
-
Nervi: Hyopmielinizzazione
-
Ridotti numero degli assoni mielinizzati: 50% del normale
-
Dimensioni di rimanenti assoni mielinizzati: 2 a 6 μm
-
Assonale sheath: Sottile per assoni diametro
-
Bulbi a cipolla: Rare
-
Cluster rigenerati: Rare
-
Tomaculae: nessuna
-
Muscoli: Lieve denervazione cambiamenti
Ipomielinizzante congenita neuropatie: Altro
Ereditarietà relapsing thermosensitive neuropatia
l
Autosomica dominante
-
Insorgenza: 6 a 43 anni (Solitamente < 15)
-
Ricorrente
-
Precipitati da febbre > 38 °C
-
2 a 5 attacchi
-
Esame normale fra attacchi
-
? Durata dei sintomi correlata con durata di febbre
-
Debolezza: Paraparesi o quadriparesis
-
Sensorio: Parestesie e intorpidimento
-
Occasionali: Disfagia; Dysuria
-
Elettrofisiologia: Demielinizzanti
-
Blocco di conduzione
-
Latenza distale prolungato
Letale Neonatale moto-sensorio Polyneuropathy1
l
Autosomica recessiva
-
Clinica
-
Insorgenza: Prenatal con ridotta movimenti fetali
-
Ipotonia
-
Debolezza
-
Distale > prossimale
-
Simmetrica
-
Diaframmatici: Insorgenza nascita to 6 mesi
-
Sensorio: Dolore ridotta in alcuni pazienti
-
Contratture (75%)
-
CNS: ? Cambiamenti correlate a ipossia
-
Prognosi
-
Aumento disabilità
-
Tarda Statico fase senza recupero
-
Morte < 8 mesi 2° Insufficienza respiratoria
-
Laboratorio
-
Elettrofisiologia
-
NCV: Ridotti o assente CMAPs e SNAPs
-
EMG: Denervazione
-
Tronco cerebrale evocata potenziali: Ridotti onde 5 suggerisce perdita di udito sensorioneurale
-
Patologia dei nervi
-
Perdita di sensorio e motor, grossi e medium di dimensione, assoni mielinizzati
-
Patologia muscolare: Atrofia a gruppi; Molte fibre sottili
-
Esclusa la
Infantile Assonale Polineuropatia con Respiratorio
Insufficienza5
l
Recessiva; Sporadica
-
Epidemiologia: Maschi > femmine
-
Clinica
-
Laboratorio
-
Elettrofisiologia
-
NCV: Ridotti o assente CMAPs e SNAPs
-
EMG: Denervazione
-
Fibrillazione
-
Alcune ingrossati Unità motorie firing a rapida rates
-
Brainsten evocata potenziali: Ridotti onde 5 suggerisce perdita di udito sensorioneurale
-
CSF: Normale
-
Patologia dei nervi
-
Perdita di sensorio e motor, grossi e medium di dimensione, assoni mielinizzati
-
Normale mielinizzazione on rimanenti assoni
-
Ultrastrutture: Büngner bands; Normale non mielinizzati assoni
-
Patologia muscolare
-
Atrofia a gruppi: Fibre sottili angular
-
grossir Fibre muscolari: Ipertrofia; Predominanza di tipo I in alcuni
-
Midollo spinale: Lieve perdita di anteriori horn cellule
-
Esclusa la
Congenita Catarrata, facciale Dysmorphism e Neuropatia Sindrome (CCFDN)2
l
CTDP1
; Cromosoma 18q23-qter; Recessiva
Navajo neuroepatopatia
l
Autosomica recessiva
-
Epidemiologia
-
Navajo: Fino ai 1 caso/1,600 nati vivi
-
Specialmente Occidente reservation
-
Insorgenza: 1 anno
-
Polineuropatia
-
Perdita sensoria: Distale; Panmodale; Acromutilazione
-
Debolezza: Distale > prossimale; Progressiva; Ritardo nel camminare
-
Riflessi tendinei: Ridotti
-
Patologia dei nervi: Gravi perdita di grossi e assoni sottili mielinizzati
-
CNS: Progressiva materia bianca lesioni alla MRI
-
Sistemico
-
Scheletro: Contratture; Bassa statura; Scoliosi
-
Epatico
-
Clinica: Epatomegalia; Reyes-episodi simili (70%)
-
Patologia: Macronodular cirrosi; Citocromo ossidasi negative epatociti
-
Oculare: Ulcerazioni cornee
-
Sistemico infezioni (80%)
-
Sessuale infantilism
-
Scarsa aumento di peso
-
Prognosi: Morte by Adolescenti in 40%
-
Laboratorio
Navajo sensorio-autonomo neuropatia con artropatia
-
Epidemiologia
-
8 bambini in 3 Navajo famiglie
-
Polineuropatia
-
Motorio: Normale
-
Perdita sensoria
-
Lieve perdita di profonda dolore e temperature sensazione
-
Corneal insensibilità
-
Alcuni pazienti con normale esame
-
Autonomo
-
Infantile febbre (Ricorrente)
-
Intolleranza al calore
-
Sudorazione: ridotta
-
Riflessi tendinei: Presente
-
Elettrodiagnostica: Spesso normale
-
Patologia dei nervi: Ridotti Sottili mielinizzati e non mielinizzati assoni
-
Scheletro: Senza dolore fratture; Progressiva artropatia
-
Cute: Ispessito on mani
-
Progressione: Solo di articolazioni e ossa Malattie
SOX10 mutazione: Ipomielinazione nel CNS e PNS;
Ipopigmentazione e Enteric aganglionosis3
l
SOX10
;
Cromosoma 22q13; Dominante o sporadica
-
SOX10 gene mutaziones12
-
Mutazioni specifiche: 12 paia di basi delezione nell'esone 5; Q250Stop; 759delG
-
Produce intatti proteina con estese tail aggiunsero to transactivation dominio nel terminale C
-
Mutazioni nulle nello stesso gene produce
-
Dominante megacolon sindrome
-
Waardenburg-Shah sindrome
:
intestinale aganglionosis; Pigmentazione difetti; Sordità, neurosensoria
-
Meccanismi della malattia
-
Gravi Malattie: ? Proteina mutata agisce come dominante negative
-
Haploinsufficiency: Fenotipo più lieve Malattie
-
SOX10 proteina
-
Clinica
-
Insorgenza
-
CNS
-
Ritardo nello sviluppo: Gravi
-
Spastic quadriplegia
-
Attacchi epilettici
-
Sordità: Sensorioneurale
-
MRI: Assenti mielinizzazione in materia bianca; Hyperintensity on T2
-
CSF: Proteina lievemente aumentati
-
Patologia: Assenti centrale mielina
-
PNS
-
Atrofia muscolare
-
? Perdita sensoria
-
Riflessi tendinei: Ridotti
-
Elettrofisiologia: Neuropatia demielinizzante
-
Patologia del nervo
-
grossi assoni: Assenti o ridotta mielina; Normale numero; Occasionali excessively grossi assoni
-
Sottili assoni: Ridotti numero
-
Cellule di Schwann: Presente; Aligned con assoni in 1 a 1 relazione
-
Oculare
-
Nistagmo
-
Atrofia ottica
-
Dystopia canthorum
-
Gastrointestinale
-
Pigmentazione: Eterocromia iridis
-
Decorso
-
? Non-progressivo
-
Morte a 3 mesi in 1 paziente
Tirosinemia
l
Fumarylacetoacetase
;
Cromosoma 15q23-q25; Recessiva
Chediak-Higashi
l
CHS1 (LYST)
;
Cromosoma 1q42.1-q42.2; Recessiva
-
CHS1 proteina
-
Funzioni
-
Interagisce con N-ethylmaleimide-sensibile fattore (NSF) attachment proteina recettori (SNAREs)
-
Ruolo nella membrana fusione o fissione
-
Può regulate lisosomi movimentazione
-
Localizzazione subcellulare: ? Citoplasmica
-
Tessuto specificità: Timo; Sangue periferico leucociti; Midollo osseo; Cervello
-
Clinica
-
Polineuropatia
-
CNS
-
Ritardo mentale
-
Spinocerebellar degenerazione
-
Insorgenza adulta: Parkinsonismo; Demenza
-
Pigmentazione difettose (Parziale albinism)
-
Cute: Giganti melanosomes; Ipopigmentazione
-
Oculare: Nistagmo; Fotofobia
-
Sistemico
-
Organomegalia: Fegato; Milza; Lymph nodi
-
Ematologici
-
Anemia; Pancitopenia
-
Emorragia: Piastrine mancanza dense granuli; funzione è anormale
-
Ridotti naturale killer cellule e Citotossica T lymphocyte funzione
-
Anormale fusione di lisosomi
-
Difettoso granule killing
-
Midollo osseo
-
Eosinofile, perOssidasi-positive inclusione corpi in myeloblasts e promyelocytes
-
Collegamento esterno: Nuove Messico
-
Lymphoreticular (Lymphohistiocytic) maligne (90%)
-
Suscettibilità a infezione
-
Eterozigoti: granulare anomaly di linfociti
-
Decorso
-
Morte: Spesso < 7 anni
-
Se maligne è assente
-
Sopravvivenza più lunga
-
Progressiva malattia neurologica
-
Patologia: Giganti lisosomale inclusioni nel CNS, Cellule di Schwann e Leucociti
-
Modello animale: Beige topo
Cerebrali Disgenesi, Neuropatia, Ichthyosis, e Palmoplantar
CHERATODERMA (CEDNIK)16
l SNAP29
; Cromosoma 22q11.2;
Recessive
-
Epidemiologia: arabe Muslim famiglie dal northern Israel
-
Genetica
-
Mutazione: 1 bp delezione; 220delG
-
SNAP29 proteina
-
Modulator di vescicole movimentazione
-
Legami alla syntaxin fusione proteine
-
Malattia: Livelli ridotta in tessuti
-
Altre mutazioni nelle vescicole riciclaggio geni
-
Clinica
-
Insorgenza
-
>Età: < 4 mesi
-
>Roving occhio movimenti
-
>Scarsa Capo e tronco controllo
-
>Mancata crescita
-
Motorio
-
>Ritardato sviluppo
-
>Tarda seduta e camminanti
-
Polineuropatia
-
>Clinica: Non ben descritti
-
>Riflessi tendinei: Assenza
-
>Elettrofisiologia: Perdita assonale
-
>Biopsie muscolari: Denervazione; Normale mitocondriale ossidativa
enzimi
-
CNS
-
>Microcefalia: Progressive
-
>Psicomotorio ritardo: Grave
-
Faccia: Dysmorphism
-
>Alpolmoneati faces
-
>Antimongolian occhio slant
-
>Ipertelorismo: Lieve
-
>Piatti larga nasal radice
-
Cute: Cheratoderma palmoplantare e Ichthyosis
-
>Insorgenza: fra 5 e 11 mesi
-
>Progressive durante 2a anno di vita
-
Occhi
-
>Ottiche disk: Ipoplastici
-
>Elettrofisiologia: Ridotta conduttanza dal periferici retina e
maculari atrofia
-
Orecchie: Sordità sensorioneurale, lieve
-
Decorso: Morte in 40% dovuta a polmoniti a all'età di 5 a 12 anni
-
Laboratorio
-
MRI del cervello
-
>Corpo calloso anormalità
-
>Corticale displasia: Pachygyria e Polimicrogyria
-
Cute
-
>Ultrastrutture: Chiari vescicole in spinous e granular epidermici
layers
-
>Glucosylceramides: Retained, ANORMALEmente, entro cellule di più bassa
stratum corneum
Ipomielinizzazione e Congenita cataratta
17
l Hyccin (DRCTNNB1A)
; Cromosoma 7p21.3–p15.3;
Recessive
-
Epidemiologia: 4 famiglie
-
Mutazioni
-
Tipo: Stop, Sito di splicing e missenso
-
Tutte omozigote
-
Hyccin proteina
-
Ubiquitously espresso in encefalo
-
Cell localizzazione: Membrana plasmatica, non uniforme
-
Effetti delle mutazioni
-
>Assenza o livelli ridotti di Hyccin proteina
-
>Mildest paziente, senza neuropatia, aveva residua proteina
-
Insorgenza
-
Età: nascita o in 1a 2 mesi di vita
-
Catarrata
-
Clinica
-
Psicomotorio
-
>Nascita: Normale
-
>1 anno: Ritardato
-
>Camminare solo con supporto
-
- Insorgenza: 1 a 2 anni
-
- Perdita: 8 a 9 anni
-
>Deficienza mentale: Lieve to Moderate
-
Piramidale: Progressive disfunzione
-
Cerebellare: Progressive disfunzione
-
Muscoli
-
>Debolezza: gambe > braccia
-
>Degenerazione
-
Occhi: Catatacts (100%)
-
>Bilaterale
-
>Chirurgia spesso condotti
-
Attacchi epilettici (30%)
-
Scoliosi (40%)
-
Survival
-
>La maggior parte un minimo to 3a decade
-
>1 morte in 1a decade
-
Laboratorio
-
MRI del cervello: Diffuse cerebrale hypomielinizzazione
-
Elettrofisiologia: Polineuropatia (90%)
-
>Motorio velocità di conduzione nervosa: Leggermente to marcatamente abbassata
dipendenti sulla età
-
>CMAP ampiezza: Ridotta
-
Biopsia dei nervi surali
-
>Carenza di fogli mielinici nei numerosi nervo fibre
-
>Perdita assonale: Lieve
-
>No attiva assonali degenerazione
Infanzia sindrome del tunnel carpale: differenziale daignosis
-
Malattie da accumulo: Mucopolysaccharidoses
;
Mucolipidoses
-
Strutturali anomalie
-
Local: Ossa; Tendinei; Muscoli; Band; Transverse carpal ligament Ipertrofia
-
Generale: Scheletro (Spondyloepiphyseal) Displasia; Weill-Marchesani
;
Melorheostosis
-
sanguinolenta: Hemophilia
-
Mass: Hemangioma; Hamartoma; Tumori; Abscesses
-
Sistemico malattia: disseminata angiomatosis; Lipoid proteinosis; Scleroderma
-
Local lesione
-
Overuse: Flessione-extension; Sostenuta flessione (Distonia; Paralisi cerebrale)
-
Trauma; Ustioni
-
Familiare
-
Comunemente autosomica dominante
-
Insorgenza: 1 anno to 3a decade
-
Frequently bilaterale o destra Mani
-
Associata con ispessiti transverse carpal ligament, o digital
flessori tenosynovitis
Sensorio ereditaria Neuropatie
Ritorno a
Neuromuscolare home page
Ritorno a
Indice delle polineuropatie
1. Muscoli Nervi 1998;21:1473-1477, 2001;24:760-768
2. Ann Neurol 1999;45:742-750, Nature Genetica 2003; Online Sept
3. Ann Neurol 1999;46:313-318
4. Malattie neuromuscolari 2000;10:592-598
5. J Bambino Neurol 2000;15:513-517
6. Malattie neuromuscolari 2001;11:395-399
7. J Bambino Neurol 2001;16:642-644
8. Acta Neuropathol 2001;Online giugno
9. Hum Molec Genet 2001;10:2783-2795
10. Neuropathol Appl Neurobiol 2002;28:170
11. Hepatology 2001;34:116-120
12. Ann Neurol 2002;52:836–842
13. J Med Genet 2002;39:838–843
14. Am J Hum Genet 2003; Ottobre, Arch Neurol 1999;56:1283-1288
15. Eur J Ped 2000;159:300-301
16.
Am J Hum Genet 2005;77: On-line giugno
17.
Nat Genet 2006;38:1111-1113
2 ottobre 2006
Va a Acronimi e sigle
Va a Mitolario in
italiano
Va a Mitolario italiano -
inglese
Va a Mitolario inglese -
italiano
{kind=link}