CMT 2C (HMSN 2C)
 27
27
l Cromosoma 12q23-q24:
Dominante
-
Caratteristiche cliniche
-
Insorgenza: Precoce 1a decade
-
Debolezza
-
Distale: Mani e piedi
-
Diaframmatici e Intercostal paralisi: Shortness di respiro
-
Corde vocali: alterato
voice; Possono essere solo segni in lievemente affetti pazienti
-
Può progredire to prossimale e faccia muscoli
-
Perdita sensoria: Asintomatica; Ridotta Vibrazioni sensi
-
Riflessi tendinei: Depressed o assenti
-
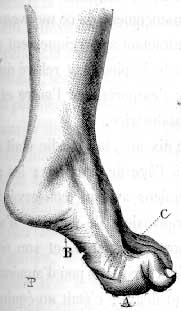
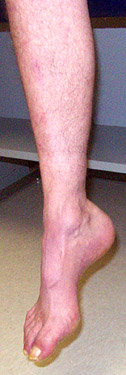
Scheletro: Pes cavus (70%); Artrogriposi in alcuni pazienti
-
Elettrodiagnostica
-
Median NCV > 50 M/s
-
CMAP ampiezzas spesso ridotta
-
Phrenic CMAP: ampiezza ridotta
-
SNAPs: Normale o ampiezza ridotta
-
EMG: Denervazione cronica
CMT 2D
l Glycyl tRNA
sintetasi
; Cromosoma 7p14; Dominante
-
Genetica
-
Epidemiologia: 4 o 5 famiglie descritti; North American, Bulgarian,
Mongolian
-
GARS proteina
-
Famiglia: Aminoacil tRNA sintetasi
-
Funzione: Charge tRNAs con cognate aminoacido (glicina)
-
Espressione: Ubiquitous
-
Altro malattie associazione: anticorpi bersglio (EJ) in
immune miopatia
-
Caratteristiche cliniche
-
Insorgenza: 16 to 30 anni; Mani debolezza
-
Debolezza
-
Distale
-
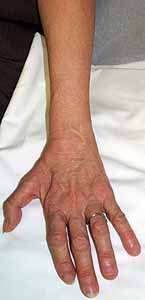
braccia > gambe: Specialmente thenar e 1a dorsale interosseus
-
Bilaterale
-
Perdita sensoria
-
Variabili: Sensazioni normale in alcuni Famiglia membri
-
Pattern: Distale; braccia e gambe; Pansensorio
-
Riflessi tendinei: Assenza o ridotta braccia; Diminuita gambe
-
Progressione: Lenta nel corso di anni
-
Elettrodiagnostica
-
Perdita assonale: Mani
-
Normale NCV
CMT 1F39
l Neurofilamenti catena sottile
; Cromosoma 8p21; Dominante o sporadica
-
Genetica
-
Mutazioni: Missenso e Delezioni
-
Capo dominio: Glu7Lys; Pro8Arg; Pro8Gln; Pro8Leu; Glu89Lys
-
Coil 1A: Asn97Ser
-
Coil 2B: Gln333Pro
-
Tail: Glu528del
-
Allelica con: CMT 2E
-
Insorgenza
-
Meno di 13 anni
-
2a decade: Pro8Arg; Glu528del
-
Prima infanzia: Altre mutazioni
-
Disturbi dell'andatura
-
Clinica
-
Weakiness
-
gambe e braccia
-
Distale > prossimale
-
Possono essere gravi
-
Precoce: Ritardato motor pietra miliare o andatura malattie
-
Muscoli atrofia
-
Riflessi tendinei: Spesso ridotta o assenti
-
Perdita sensoria
-
Tremore: Alcuni pazienti
-
Elettrofisiologia
-
NCV motoria: 15 a 38 M/s
-
SNAPs: Spesso assenti
-
Patologia dei nervi (1 paziente)
-
Perdita assonale: grossi e non mielinizzati assoni
-
rigeneranti insiemi
-
Bulbi a cipolla (Sottili)
-
Sottili mielinizzati assoni
CMT 2E4
l Neurofilamenti catena sottile
; Cromosoma 8p21; Dominante
-
Genetica
-
Mutazioni
-
Tipi
-
Missenso
-
Delezione: In frame e frame deriva
-
Localizzazioni
-
Solitamente in Capo e bastoncelli domini
-
Thr21fs; Pro22Ser; Leu268Pro; del322Cys_326Asn; Gln333Pro;
Glu396Lys
-
Effetto della mutazione: ? Alters auto assemblaggio di NFL33
-
Haploinsufficiency: Probabilmente non patogenico
-
Allelica con: CMT 1F
-
Neurofilamenti catena sottile proteina
-
Richiesta per organizzazione di neurofilamenti
-
Associata con radiale assonali crescita
-
Epidemiologia: multipla famiglie
-
Insorgenza
-
Età: 1a to 5o decade
-
Difficoltà camminanti; Debolezza nelle gambe
-
Caratteristiche cliniche
-
Neuropatia
-
Debolezza
-
Perdita sensoria
-
Distale
-
gambe > braccia
-
Pansensorio, specialmente vibrazione
-
Sensoria Atassia (20%)
-
Riflessi tendinei: Ridotta o assenza
-
Atassia episodica: In fanciullezza; Scatenati da febbre
-
Progressione: Lenta nel corso di anni; Alcune gravi, altri lieve,
disabilità
-
Perdita di udito (30%)
-
Postural tremore (30%)
-
Sistemico caratteristiche
-
Pes cavus: 100% over 20 anni di età
-
Qualche paziente con hyperkeratosis
-
Elettrodiagnostica: Studi di conduzione nervosa
-
Perdita assonale: Sottili CMAP
-
NCV: Normale o leggermente ridotta; Median - Gamma di 29 to 55 M/s
-
Latenza distale: Possono essere prolungato
-
tronco cerebrale uditive evocata potenziali: anormale
-
MRI di encefalo e midollo spinale: Normale
-
Patologia dei nervi48
-
Giganti assoni
-
Filled con densely packed neurofilamenti
-
Surrounded by thnella fogli mielinici
-
Non presenti con Glu396Lys mutazione
-
Assonale atrofia: Alcune regioni senza neurofilamenti
-
rigeneranti insiemi
-
Demielinizzazione: Secondaria; Occasionale sottili bulbi a cipolla
-
Similarities to:
Neuropatia con assoni giganti
CMT 2F19
l HSPB1 (HSP 27)
; Cromosoma 7q11-q21;
Dominante
-
Epidemiologia: russe (Voronezh) e famiglie belghe
-
Genetica
-
Insorgenza: 15 a 25 anni
-
Clinica
-
Debolezza
-
Distale; gambe > braccia; Simmetrica
-
Possono essere gravi con piedi cadenti
-
Riflessi tendinei: Ridotta
-
Perdita sensoria: Lieve
-
Distale
-
Piedi e Mani
-
Specialmente dolore e temperature
-
Fascicolazioni: In anziano pazienti
-
Nervi craniali: Normale
-
CNS: Normale
-
Progressione
-
Lenta
-
Disabilità nella maggior parte delle dopo 15 a 20 anni
-
Alcuni pazienti rimangono ambulante dopo 30 anni
-
Laboratorio
-
Elettrodiagnostica: Perdita assonale
-
CMAPs: Sottili o assenza; 30% a 50% nell'uomoi; 10% del normale o
unobtainable in piedi
-
NCV: 42 a 58 M/s
-
EMG: Denervazione distale
CMT 2G46
l Cromosoma
12q12–q13.3; Dominante
-
Epidemiologia: spagnoli Famiglia
-
Genetica: Locus simile to CMT 4H
-
Insorgenza
-
Età: 2a decade; Gamma 9 to 76 anni
-
Deformità nei piedi
-
Difficoltà camminanti
-
Clinica
-
Debolezza: gambe > braccia
-
Perdita sensoria: gambe; Distale
-
Riflessi tendinei: Ridotta a caviglie
-
Deformità nei piedi
-
Decorso: Lentamente progressive
-
Laboratorio
-
Elettrodiagnostica
-
Motorio
-
NCV: Normale o leggermente abbassata
-
CMAPs: Sottili nelle gambe
-
SNAPs: Piccola ampiezza nelle gambe
-
EMG: Denervazione distale nelle gambe
-
Patologia
-
Perdita assonale: Distale > prossimale; Rigenerazione
-
Cellule del corno anteriore: Ridotta numero
-
Dorsal radice cellule gangliali: Ridotta numero
CMT 2L47
l HSPB8
; Cromosoma 12q24-qter;
Dominante
-
Genetica
-
Epidemiologia: 1 Famiglia cinese
-
Insorgenza
-
Età: 15 a 33 anni
-
Debolezza nelle gambe
-
Clinica
-
Debolezza
-
Distale: 100%
-
gambe: 100%
-
Prossimale: Qualche paziente
-
Mani: 30%
-
Simmetrica
-
Perdita sensoria
-
Distale
-
gambe
-
No indolore lesione o ulceration
-
Riflessi tendinei: Ridotta o assenza
-
Scheletro
-
Pes cavus 80%
-
Scoliosi 15%
-
Decorso
-
Progressione: Lenta
-
mai carrozzina dipendenti
-
Laboratorio
-
Elettrofisiologia
-
NCV: Normale
-
SNAPs: Assenza o Sottili ampiezzas
-
EMG: Denervazione cronica
-
Patologia dei nervi
-
Perdita assonale
-
Rigenerazione
HMSN-P
CMT 2-P0
Ereditarie Neuropatia motorio-sensoria con Atassia26
l Cromosoma 7q22-q32;
Dominante
-
Epidemiologia: Singola American Famiglia di irlandesi ancestry
-
Insorgenza
-
Età: 13 a 27 anni; No anticipazione
-
Disturbi dell'andatura, specialmente in scuro
-
Clinica: Variabili espressione
-
Atassia: Dismetria; Ataxic andatura; Nistagmo
-
Perdita sensoria: Vibrazioni > Propriocezione; Positive Romberg
-
Riflessi tendinei: Ridotta o assenza
-
Estensore plantar riflessi: 50%
-
Debolezza: Distale o Prossimale; gambe o braccia; 70%; Più a più vecchia all'età di
-
Scheletro: Pes cavus; dita a martello
-
No demenza
-
Progressione: Lenta; Normale vita span
-
Laboratorio
-
NCV: Neuropatia assonale; Sottili o assenti SNAPs; Occasionale prolungato
distale latenza
-
EMG: Denervazione cronica
-
MRI: Atrofia cerebellare, lieve, nei pazienti più vecchi
CMT con intermediate NCV (DI-CMT)17
DI-CMT, Tipo A
l Cromosoma
10q24.1-q25.1; Dominante
-
Epidemiologia
-
Insorgenza: 1a o 2a decade
-
Clinica
-
Debolezza
-
Distale
-
gambe e braccia
-
Simmetrica
-
più grave by 4a e 5o decade
-
Perdita sensoria: Distale; Lieve
-
Riflessi tendinei: Assenza
-
Nervi craniali: Normale
-
Autonomo funzioni: Normale
-
Progressione: Lenta over decadi; Più in 5o decade; Molte mai use
carrozzina
-
Laboratorio
-
Elettrodiagnostica
-
CMAPs: Sottili o assenza
-
NCV: 25 a 45 M/s
-
SNAPs: Basso ampiezza
-
CSF proteina: Normale, o Lievemente aumentato
-
Patologia
-
Perdita di grossi mielinizzati assoni
-
Bulbi a cipolla: Occasionale on EM
DI-CMT, Tipo B
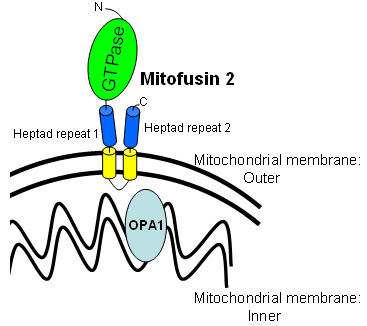
l Dinamina 2 (DNM2)
; Cromosoma 19p12-p13.2;
Dominante
-
Epidemiologia
-
australiane, Belgium e nordamerica famiglie
-
DNM2 genetica
-
DNM2 proteina
-
Famiglia di grossi GTPases
-
Parte di cellulare fusione-fissione apparato
-
Insorgenza: 1a o 2a decade
-
Clinica
-
Debolezza: Distale; gambe e braccia; Simmetrica
-
Perdita sensoria: Distale
-
Tremore
-
Nervi craniali: Normale
-
Autonomo funzioni: Normale
-
Progressione
-
Lenta over decadi
-
Molte mai use carrozzina
-
Sistemico: Neutropenia (Lys558 mutazioni)
-
Laboratorio
-
Elettrodiagnostica
-
CMAPs: Sottili o assenza
-
NCV: 24 a 54 M/s
-
SNAPs: Basso ampiezza
-
CSF proteina: Normale, o Lievemente aumentato
-
Patologia
-
Perdita di grossi mielinizzati assoni
-
Bulbi a cipolla: Occasionale
DI-CMT, Tipo C43
l tyrosyl-tRNA
sintetasi (YARS)
; Cromosoma 1p34-p35;
Dominante
-
Epidemiologia
-
2 Familie: Midwestern US (tedesche); Bulgarian
-
genetica59
-
Mutazioni: Missenso (G41R e E196K); Delezione (In frame
153-156delVKQV)
-
Glycyl-tRNA sintetasi mutazioni in CMT 2D
-
Proteina
-
Normale
-
Funzione: Catalizza aminoAcilazione di tRNAtyr con
tirosina
-
Espressione: Ubiquitous, includono encefalo e midollo spinale
-
Concentrata in granular struttura in crescita coni, branch
Puntiformes e distale neurites
-
mutante: Parziale perdita dell'attività; Possono avere effetto dominante negativo
on funzione del normale proteina
-
Insorgenza: Gamma 7 a 59 anni; La maggior parte in 1a o 2a decade
-
Clinica
-
Debolezza: Distale; gambe > braccia; Simmetrica
-
Perdita sensoria: Distale; Lieve
-
Nervi craniali: Normale
-
Autonomo funzioni: Normale
-
Progressione: Lenta over decadi; Molte mai use carrozzina
-
? Meno gravi nelle femmine
-
Laboratorio
-
Elettrodiagnostica
-
Patologia
-
Perdita di grossi mielinizzati assoni: Più con l'aumento dell'età
-
rigeneranti insiemi
-
No bulbi a cipolla
Altro CMT malattie con intermediate NCV
Recessive, CMT assonale
CMT assonale (AR-CMT2A; CMT2B1)1
l Lamina A/C
; Cromosoma 1q21.2
-
Epidemiologia: nordafricani famiglie
-
Genetica
-
Mutazione: R298C; omozigote
-
Allelica con
-
Emery-Driefuss distrofia muscolare 2
-
Autosomica dominante cardiomiopatia dilatativa con A-V blocco (CMD1A)
-
Mutazioni in bastoncelli dominio
-
Familiare Parziale LIPODISTROFIA (Köbberling-Dunnigan Sindrome)
-
Mutazioni in globular Terminale C dominio
-
LGMD 1B
-
Mandibuloacral displasia
:
-
Mutazione: R527H (Terminale C dominio), Recessive
-
Possono essere Associata con Parziale LIPODISTROFIA
-
Atrial fibrillazione: Insorgenza precoce; Missenso (E161K) mutazione10
-
quadricipiti miopatia con la cardiomiopatia dilatativa: Missenso
(R377H) mutazione
-
Premature invecchiamento
-
Hutchinson–Gilford progeria
: Recessive;
anormale nucleare architettura
-
Werner’s sindrome, atipiche
: Dominante;
più grave
-
Insorgenza
-
Età: 2a decade; Gamma 4 a 24
-
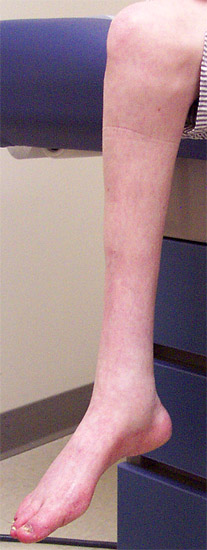
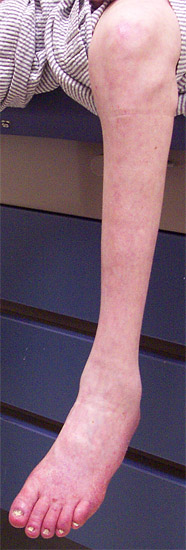
Segni: Debolezza distale, specialmente gambe
-
Clinica
-
Debolezza
-
Arti
-
Distale Early
-
Progressione to prossimale coinvolgimento nelle gambe (60%)
-
gambe > braccia
-
Simmetrica
-
Progressione
-
Rapido over 4 anni
-
Pronunciato disabilità
-
Deperimento muscolare: Distale
-
Riflessi tendinei: Ridotta in più bassa estremità
-
Perdita sensoria
-
Distale
-
Mani e piedi
-
Modalità: Variabili, specialmente grossi fibre
-
Peggiora con malattie progressione
-
Scheletro
-
Pes cavus (80%)
-
Cifoscoliosi (30%): con debolezza prossimale
-
Laboratorio
-
Nervi conduzione : Perdita assonale
-
Velocità normale
-
SNAPs: spesso assenti
-
Biopsia dei nervi
-
Assoni mielinizzati: Ridotta, specialmente assoni grossi
-
Assoni non mielinizzati: Ridotta
-
Rigenerazione: Lieve
CMT assonale (AR-CMT2B; CMT2B2)12
l Cromosoma 19q13.3;
Recessive
-
Epidemiologia: Costa Rican Famiglia, spagnoli ancestry
-
Insorgenza
-
Età: 26 to 42 anni; Media 32 anni
-
Debolezza: Simmetrica; Ankles ± Mani (66%)
-
Clinica
-
Debolezza
-
Distale
-
Simmetrica
-
gambe (100%): Intrinseca piedi; Peroneal; Calves; Anteriori tibial
-
Mani (80%): Intrinseca muscoli; dito extension; Wrist Flessione
-
Perdita sensoria
-
Distale
-
Piedi e Mani
-
Tipo
-
Panmodale
-
Fibre grosse Modalità : Specialmente coinvolta
-
Sensoria Atassia
-
Riflessi tendinei
-
Spesso assenti a caviglie
-
Possono essere ridotta o assenti altrove
-
Sofferenza
-
Crampi: Distale
-
parestesie: Distale
-
Autonomo: Normale
-
Nervi craniali: Normale
-
Decorso
-
Lentamente progressive
-
La maggior parte rimangono ambulatorio con o senza cane
-
Scheletro: Normale o Lieve deformità dei piedi
-
Laboratorio
-
Nervi conduzione : Perdita assonale
-
Motorio
-
Velocità di conduzione: Lievemente ridotta (30 a 65 M/s in braccia)
-
CMAP: Sottili o assenti nelle gambe e braccia
-
Sensoria
-
Assenza sural SNAPs
-
ampiezza ridotta SNAPs in braccia
-
Velocità di conduzione: Ridotta
-
EMG: Denervazione distale
CMT assonale (AR-CMT) con hoarseness23
l gangliosidi-indotte
differenziazione-Associata proteina 1 (GDA P1)
 ; Cromosoma 8q21.1; Recessive
; Cromosoma 8q21.1; Recessive
-
Nosologia
-
Called CMT 2K by OMIM
-
CMT RIA: Conduzioni dei nervi intermedie
-
Genetica
-
Mutazioni: Codoni di stop, o nonsenso mutazioni
-
Stesso gene come recessiva, demielinizzanti CMT 4A
-
Simile locus to AR-CMT con piramidali
coinvolgimento
-
Stessa mutazioni Possono produrre demielinizzanti o assonali fenotipo : S194X
-
GDA P1 protein52
-
Espressa in: Neuroni (DRG) > cellule di Schwann; CNS (Cervello e midollo spinale)
-
Localizzazione subcellulare:
I mitocondri, localizzata dal terminale C dominio transmembranico
-
Strutturali Famiglia: Sequenza similarità to Glutathione
S-trasferasi ma no GST attività
-
Epidemiologia: spagnoli famiglie
-
Insorgenza
-
Età: Nascita a 2 anni
-
Ipotonia
-
Piedi: DeFormity; Debolezza
-
Clinica
-
Paralisi delle corde vocali
-
Insorgenza: 2a decade
-
Raucedine (80%)
-
Non presenti in Famiglia con S194X mutazione
-
Altro nervi craniali: Normale
-
Debolezza (100%)
-
Distale: Grave; Mani e piedi
-
Prossimale: Moderate; gambe > braccia
-
Perdita sensoria (100%): Distale; Mani e piedi
-
Riflessi tendinei: Assenza
-
Progressione: Disabilità by fine di 1a decade
-
Nervi conduzione esaminazione
-
CMAPs: Ridotta o assenti
-
NCV: Normale o lievemente ridotta in prossimale nervi; 40 a 50 M/s
-
SNAPs: Ridotta o assenti
-
CSF proteina: Normale
-
Biopsia dei nervi
-
Perdita di mielinizzati assoni: Specialmente grossi, Nessuno > 7μM
-
Rigenerazione assonale e atrofia, occasionali degenerazione
-
No demielinizzazione
-
Occasionale bulbi a cipolla
-
Assoni non mielinizzati: Aumentati; Probabilmente in rigenerazione
-
Vedi anche: AR-CMT Ouvrier tipo
CMT assonale (AR-CMT): Ouvrier tipo
l autosomica recessiva
-
Insorgenza: Prima infanzia
-
Debolezza: Distale; Simmetrica; gambe prima braccia
-
Perdita sensoria: Lieve
-
Progressione: Lenta; Grave Debolezza distale by 20 anni
-
Patologia: Perdita assonale
-
Vedi anche: AR-CMT con raucedine
CMT assonale con piramidali coinvolgimento (AR-CMT2C; CMT4C2)14
l Cromosoma 8q21.3;
Recessive
-
Genetica: Simile locus to AR-CMT con raucedine
-
Epidemiologia: Tunisian Famiglia
-
Insorgenza: 4 a 8 anni; Difficoltà camminanti
-
Caratteristiche cliniche: Omogeneità in Famiglia
-
Debolezza e degenerazione: Distale; gambe poi braccia
-
Perdita sensoria: Distale
-
Riflessi tendinei: Vivaci, eccetto assenti a caviglie
-
Altro riflessi: + Hoffman e Palmo-mentale
-
Decorso: Progressive
-
Laboratorio
-
Elettrodiagnostica: Perdita assonale
-
Biopsia dei nervi: Grave perdita assonale; Occasionale rigenerazione
CMT assonale con acrodystrophy
l Recessive8
-
Epidemiologia: turche Famiglia, consanguinei
-
Clinica
-
Insorgenza: 7 a 10 anni; Senza dolore ulcere on piedi
-
Debolezza: Distale > prossimale; Simmetrica; Mani e piedi
-
Degenerazione: Distale
-
Perdita sensoria: Distale; Simmetrica; Mani e piedi; Panmodale
-
Riflessi tendinei: Assenza
-
Acrodistrofia: Mutilazione e Ulcere on piedi e mani
-
Decorso: Lentamente progressive
-
Laboratorio
-
NCV
-
Velocità normale quando obtainable
-
Motorio e Sensoria ampiezzas: Ridotta
-
EMG: Denervazione distale
-
Patologia dei nervi
-
Assoni mielinizzati: Ridotta o assenti
-
Assoni non mielinizzati: Ridotta (30% del normale)
-
Endoneuriale collagen: Aumentati
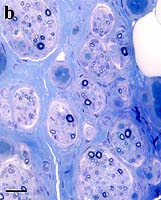
HMSN III (Dejerine-Sottas)32
-
Generale
-
Grave
-
Infantile insorgenza
-
Lentamente progressive
-
Tipi genetici: Dominante sono spesso sporadico
-
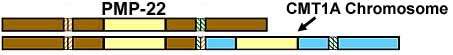
DSS-A: mutazioni puntiformi PMP-22
l Cromosoma
17; Dominante
-
Genetica
-
Localizzazione delle mutazioni
-
Transmembranali domini
-
Domini 1, 2, 3 e 4
-
Dominio transmembranico mutazioni Vedi anchen in
CMT1A)
-
Punto caldo per mutazioni: Ser72
-
26% delle mutazioni puntiformi PMP-22
-
Mutazioni: Ser72Pro; Ser72Trp; Ser72Leu (La maggior parte
frequenti)
-
Fenotipo: Solitamente gravi; Insorgenza 1a anno; La maggior parte
non camminano mai
-
Mutazione tipo: CG to TG transizioni comune (~ 40% delle mutazioni)
-
DSS-B: P0 mutazione puntiforme
l Cromosoma
1q22; Recessive o dominante
-
Genetica malattie meccanismi
-
Dominante negative
-
Missenso mutazioni nella membrana, Intracellulare, o
dominio extracellulare
-
19 mutazioni identificate
-
Transmembranali mutazioni di P0 over-rappresenta in
DSS gruppo
-
Esempi: Gly167Arg; Tyr82Cys
-
Eterozigote
-
Maggio apparire sporadically e poi transmit come Dominante
-
Altro mutaton modelli producente gravi malattie
-
Mutazioni la quale distrutto the MPZ terziaria struttura
-
La maggior parte mutazioni che truncate the citoplasmatiche MPZ dominio:
Eliminate aminoacidi 198–201 (RSTK)
-
Mutazione puntiforme alterante Ser204 (? Fosforilato via PKC
Legante al RSTK dominio)
-
omozigote assenza di P0 proteina
-
Clinica
-
Insorgenza
-
Età: 1a 2 anni
-
Tone: Ipotonico prima di 9 mesi
-
Camminare: Solitamente dopo 18 mesi
-
Riflessi tendinei: Assenza
-
Decorso
-
Camminare
-
La maggior parte dei pazienti in grado a camminare a alcuni time
-
La maggior parte dei pazienti eventualmente necessitano ambulation aids
-
Wheelchair: Alcuni pazienti
-
Progressione in debolezza: Alcuni pazienti
-
Life aspettativa
-
La maggior parte normale
-
Alcune Early morti dovuta a respiratorie failue
-
Nervi Ipertrofia: Alcuni pazienti
-
Laboratorio
-
CSF proteina: 50 a 167 mg/dL
-
Elettrofisiologia
-
Motorio velocità di conduzione nervosa: < 15 m/s in braccia
-
Non blocco di conduzione o temporale Dispersione
-
CMAP e SNAP ampiezzas: Sottili
-
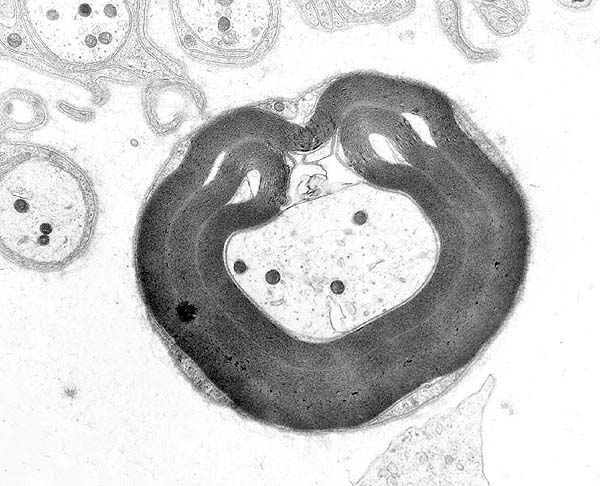
Patologia: Irregolare ripiegature e redundant anse della mielina (Tomaculae)
-
DSS-C
l Cromosoma
8q23-q24; Dominante
-
Epidemiologia: Iowa Famiglia
-
Insorgenza: 2 anni; Steppage andatura
-
Clinica
-
Debolezza: Progressive; Distale > prossimale; Mani e piedi
-
Perdita sensoria: Distale > prossimale
-
Riflessi tendinei: Assenza
-
Nervi ingrossati
-
Charcot giunture
-
CMT 4F
l Periassina;
Cromosoma 19q13; Recessive
-
Dominante
-
Perdita di udito
-
Insolita faces
-
DSS-EGR2
l Precoce Crescita
Response 2 (EGR2)
; Cromosoma
10q21.1-q22.1; Dominante, de novo
-
Mutazione: Arg359Trp
-
Caratteristiche cliniche: Vedi EGR2
-
Altro EGR2 mutazioni
-
CMT 1F
-
Dejerine-Sottas: Caratteristiche cliniche
-
Insorgenza: < 3 anni
-
Debolezza
-
Diffuse; Distale > prossimale
-
Ritardato motor pietra miliare: Camminare 15 a 48 mesi
-
Peak perFormance: 5 a 20 anni
-
Tone: Ridotta
-
Perdita sensoria: Tutte Modalità ; Grave; Distale > prossimale
-
Riflessi tendinei: Areflexic
-
Ingrandimento dei nervi
-
Alcuni pazienti: Gly138Arg mutazione31
-
Possono essere asimmetrico e multifocalei o simmetrica
-
Craniale nervo malattie: Occasionale
-
Possono essere Associata con nervi craniali ingrandimento
-
Oculare
-
Miosi
-
Pupille: Ridotta risposta alla luce
-
Ptosi
-
Nistagmo
-
Limitazioni EOM
-
Perdita di udito: Lieve
-
Morfologia: Bassa statura, Grinzosità facciale caratteristiche e Full labbra Avviene
-
Scheletro: Cifoscoliosi; Deformità nei piedi (Club piedi)
-
Progressione: Lieve fino alla teens; Grave disabilità può aversi
-
Laboratorio
-
Elettrofisiologia: Demielinizzanti + Perdita assonale
-
NCV: Molto Lenta (< 12 M/s)
-
Latenza distale: Prolungata
-
CMAPs: ampiezza ridotta
-
SNAPs: Assenza
-
CSF proteina: moderatamente alto; 70 a 200 mg/dl
-
Patologia
-
Bulbi a cipolla
-
Nervi ingrossati: Aumentati fascicular area
-
fogli mielinici
-
Ipomielinizzazione
-
lunga lengths di demieliinizzati assoni
-
Pochi macrofagi con mielina detriti
-
Assoni
-
grossi mielinizzati assoni: Ridotta numero, specialmente distalmente
-
Assoni non mielinizzati: Normale numero; Lieve riduzione in dimensione
-
Vedi anche:
Neuropatia da ipomielinizzazione congenita
-
Escludono la:
Infantile CIDP
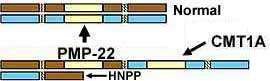
Ereditarie Responsabile to Pressione Palsies (HNPP)
l Delezione del PMP-22;
Cromosoma 17p11.2-p12; Dominante
HNPP: Genetica
-
Mutazioni
-
comune mutazione: Delezione in 85%
-
Cromosomiche delezione di regioni contenente PMP-22
-
Deleted regioni è identiche to regioni duplicato
in CMT IA
-

-
Deleted regioni contiene altre geni includono
COX10
-
Puntiforme mutaziones18:
8 identificate
-
Tipi
-
Nonsensi: Codone di stop G183A (Trp61a op), G372A (Trp124stop)
-
Frameshift: Terminazione prematura (19-20delAG, 434delT);
Più polmonehe trascrive 281-282insG
-
Sito di splicing: 78+1G>T; 179+1G>C (fenotipo lieve)
-
Missenso: Val30Met; Ala67Thr; Thr118Met65
-
Solitamente cause perdita di funzione di PMP-22
-
Database delle mutazioni puntiformi PMP-22:
IPNMDB
-
omozigote missensi mutazione
-
Thr118Met: Associata con gravi perdita assonale senza
demielinizzazione65
-
Clinica - Genetica Correlazioni
-
Penetranza: Variabili
-
Storia famigliare
-
37% dei pazienti con gene delezione non hanno Storia famigliare
-
"Sporadica" casi spesso hanno delezioni di origine poaterna.
-
Dovuta ad ineguale ricombinazione di cromosomi omologhi 17s
-
Rare materna l'origine: Dovuta a intraRiarrangiamento cromosomale
-
20% dei pazienti affetti con HNPP e delezione del PMP-22 hanno de
novo mutazione
-
16% a 30% di famiglie con HNPP fenotipo non hanno Delezione del PMP-22
-
Alcune hanno PMP-22 mutazione puntiforme
-
G-inserzione in una stiramento di sei Gs a nt 276-281
-
Alcune PMP-22 delezioni può presentarsi come cronica demielinizzanti
neuropatia mimanti CMT 1A
-
PMP-22 Proteina: Espressione ridotta in nervi in
HNPP
HNPP: Caratteristiche cliniche44
-
Insorgenza
-
Media: 19 to 26 anni
-
Gamma: 2 a 64 anni
-
Alcuni pazienti asintomatiche
-
episodi del nervo paresi
-
Insorgenza acuta
-
Sensazioni
-
parestesie
-
Intorpidimento
-
Solitamente indolore
-
Perdita può aversi senza debolezza
-
Debolezza: In distribuzione del nervo lesion
-
Prodromi
-
Lieve trauma o compression: 40%
-
ripetuta locale esercizio
-
Stiramento
-
Al risveglio: 10%
-
Chirurgia: Occasionale reports45
-
Number: 1 a 10
-
Recupero
-
Over giorni a mesi
-
Complete 50%
-
Grave lunghezza termine motor defecit 9%
-
Neuropatia caratteristiche
-
Motorio: Debolezza
-
Correlati to nervo palsie: Comunemente lieve e transitoria
-
persistente: Ulnar mani (Lieve); piedi drop (Asimmetrica)
-
Perdita sensoria
-
Vibrazioni e Dolore comunemente ridotta: Distale; Simmetrica; gambe
-
Altro regioni correlate to nervo palsie
-
Asimmetrica
-
Spesso
-
focale lesioni (62%): Correlati to stiramento, minore ripetitivo
focale trauma o pressione
-
Nervi dimensione
-
Ingranditi: Diffusamente; Visible by ultrasound
-
focale segni
-
Generale
-
Localizzazioni: A usuale siti del nervo compression
-
Variabili fra le pazienti
-
Radial: Spiral groove di humerus
-
Ulnar: Gomito
-
Median: Wrist; Isolata tunnel carpale sindrome rare
-
Peroneal: Capo di fibula
-
Plessopatia brachiale
-
Riflessi tendinei
-
Normale in 62%
-
Assenza in alcuni pazienti
-
Caviglie la maggior parte delle spesso perdita
-
Crampi: 40%
-
Scheletro
-
Pes cavus: Alcune patientsl Meno gravi o comune che con CMT1A
-
Scoliosi: Qualche paziente
-
Decorso
-
Episodica alterazioni
-
Alcune permanente debolezza Può sviluppo
-
Grave disabilità: Rare
-
Progressive Perdita di assoni: Non preminenti, eccetto Associata con
nervo compression
-
Occasionale caratteristiche
-
focale segni e perdita Sensoria a
-
Insolita siti di compression
-
Nervi craniali
-
Trigemino
-
facciale
-
Hearing loss66
-
Insorgenza: Post-natal
-
Sensorioneurale: Lieve
-
Progressive
-
Ricorrente laringe (Vocal cords)
-
Hypoglossal42
-
Progressive generalizzato moto-sensorio neuropatia
-
Può aversi senza pressione palsie
-
CMT 1 + HNPP sindrome con frame deriva G inserzione in nt
276-281
-
Possono avere insorgenza più tardi nel decorso: 5o e 6th decade
-
Occasionale casi rapidamente progressiva
-
Tremore
-
Varianti:
Davidenkow fenotipo
-
Diagnosi differenziale: Pazienti con multifocalei neuropatie senza a
17p11 delezione
-
Ricorrente neuralgic Amiotrofia
-
multipla lesioni un comune entrapment siti
-
A zone assonali neuropatia
-
PMP-22 mutazione puntiforme
HNPP: Laboratorio caratteristiche
CMT 4
Caratteristiche generali
-
Ereditarietà: Recessive
-
Demielinizzanti
CMT 4A
l gangliosidi-indotte
differenziazione-Associata proteina 1 (GDA P1)
; Cromosoma 8q21.1; Recessive
-
genetica22
-
GDA P1 protein52
-
Espressa in: Neuroni (DRG) > cellule di Schwann; CNS (Cervello e midollo spinale)
-
Localizzazione subcellulare:
I mitocondri, localizzata dal terminale C dominio transmembranico
-
Strutturali Famiglia: Sequenza similarità to Glutathione
S-trasferasi ma no GST attività
-
Epidemiologia: Tunisian famiglie
-
Insorgenza: Infantile < 2 anni
-
Debolezza
-
Rapidamente progressive
-
Distale arto: Mani e piedi
-
Grave
-
Nervi conduzione
-
Bassa velocità
-
Motorio e Sensoria: Media 30 M/s; Gamma 25 a 35 M/s
-
Patologia: Demielinizzanti
-
Mielina
-
Ipomielinizzazione
-
Segmentale demielinizzazione
-
Mielina ripiegature: Lieve o assenti
-
Bulbi a cipolla
-
Perdita di mielinizzati assoni: Specialmente grossi; A Early età
HMSN con fogli mielinici focalemente ripiegati (CMT 4B)
CMT 4B
l
Miotubularina-correlate proteina-2 (MTMR2)
; Cromosoma 11q22.1;
Recessive
-
Mutazioni
-
Tipi: Nonsensi; Frame deriva; Esone skipping delezione
-
Effetto: Ridotta fosfatasi attività
-
Miotubularina-correlate proteina-2
-
Alte livelli in
-
Neuroni
-
cellule di Schwann: mielinizzanti e Non-mielinizzanti
-
Dual specificità fosfatasi: Famiglia di 20 proteine
-
Characterized by proteina/tirosina fosfatasi e SID dominio
-
SID dominio interagisce con proteine contenente SET dominio che
associate con chromatin
-
Azione enzimatica: Defosforilazione fosfatidilinositolo 3-fosfato e
fosfatidilinositolo 3,5-bisfosfato
-
Miotubularina Famiglia malattie:
Miopatia centronucleare; CMT 4B2
-
Interagisce con: MTMR5
; Dlg1
-
geografica locazione: italiane e Arabia Sauditan famiglie
-
Caratteristiche cliniche
-
Insorgenza: < 4 anni; Media 2 anni
-
Debolezza
-
Distale e prossimale più bassa estremità
-
Simmetrica
-
Progressive: Adulti spesso carrozzina legato
-
facciale: Alcune con synkinesis
-
Perdita sensoria: Panmodale; Distale
-
Riflessi tendinei: Assenza
-
Pes equinovarus
-
Pronunciato Spessa labbra
-
Elettrofisiologia
-
Demielinizzazione: NCV 9 to 20 M/s
-
Mielinizzate Perdita di assoni
-
Patologia
-
Irregolare ripiegature e redundant anse della mielina (Tomaculae)
-
Mielinizzate Perdita di assoni
-
Modello di topo
-
anormale mielina ripiegature: Paranodal; Distale nervi > prossimale
-
Nervi conducton Velocità: Normale
-
Mutazioni
-
E276X mutazione, omozigote
-
Conditional Knock-out
-
cellule di Schwann difetti produce neuropatia
-
Neuronali difetti do non
CMT 4B2
2
l SET Legante fattore
2 (SBF2) (MTMR13)
; Cromosoma 11p15; Recessive
-
Genetica
-
Proteina
-
Famiglia: Myotubularins, pseudo-fosfatasi branch
-
Generale azioni: Phosphoinositide-mediata signalling eventi: ?
Associata con controllo di mielinizzazione
-
Miotubularina Famiglia malattie:
Miopatia centronucleare; CMT 4B
-
geografica locazione: Turchia; Tunisia; ? giapponese
-
Insorgenza: 1a 2 decadi: No Ritardato sviluppo motorio nella fanciullezza
-
Clinica
-
Debolezza
-
Insorgenza: Distale gambe; Intrinseca foot e anteriori gambe muscoli
-
Pattern: gambe > braccia; Distale; Simmetrica
-
Progressione: Debolezza prossimale in alcuni pazienti anni dopo l'insorgenza
-
Perdita di ambulation in alcuni pazienti
-
Perdita sensoria: Distale; Grave
-
Riflessi tendinei: Assenza nelle gambe; Ridotta in braccia
-
Scheletro: Pes cavus; dita a martello
-
Elettrofisiologia
-
Lenta velocità di conduzione nervosa (15 a 30 M/sec)
-
Perdita assonale: Motorio e Sensoria
-
Patologia
-
Irregolare ripiegature e redundant anse della mielina (Tomaculae)
-
Segmentale demielinizzazione
-
Bulbi a cipolla
-
Mielinizzate Perdita di assoni: grossi > Sottili
HMSN con fogli mielinici focalemente ripiegati: con giovanile-insorgenza Glaucoma6
l SET Legante fattore
2 (SBF2) (MTMR13)
; Cromosoma 11p15; Recessive
-
Allelica con: CMT 4B2
-
geografica locazione: giapponese Famiglia
-
Insorgenza
-
1a decade
-
Glaucoma
-
Debolezza; Difficoltà a correre
-
Clinica
-
Debolezza
-
Distribuzione: Distale; gambe e braccia; Simmetrica
-
Progressione: Lenta
-
Perdita sensoria: Distale; braccia e gambe; Panmodale
-
Riflessi tendinei: Assenza
-
Scheletro: Pes cavus; dita a martello
-
Intelletto: Normale
-
Oculare
-
Acuità visiva: Ridotta
-
Intraocular pressione > 21 mm Hg
-
Glaucomatous alterazioni in ottica disk
-
Laboratorio
-
CSF: Proteine alte
-
Elettrofisiologia
-
Lenta velocità di conduzione nervosa (15 a 20 M/sec)
-
Perdita assonale: Motorio e Sensoria
-
Patologia
-
Irregolare ripiegature e redundant anse della mielina
-
Segmentale demielinizzazione
-
Sottili mielinizzati assoni
-
Bulbi a cipolla
-
Mielinizzate Perdita di assoni: grossi > Sottili
HMSN con fogli mielinici focalemente ripiegati: Dominante
l P0
proteina; Cromosoma 1q22-1q23; Dominante
l ? addizionali locus
-
Fogli mielinici focalemente ripiegati
HMSN con fogli mielinici focalemente ripiegati: addizionali locus
l Recessive
CMT 4C
l SH3TC2 (KIAA1985)
; Cromosoma 5q32; Recessive
-
Epidemiologia
-
Relatively frequenti cause di AR-CMT
-
Numerose ethnic origins di famiglie
-
Sporadica pazienti identificate
-
genetica41
-
Mutazioni
-
Troncamento e missenso
-
Alcune mutazioni missenso coinvolgono splicing alterazioni: Alter sequenze
di pieno lunghezza proteina
-
Tutti i pazienti omozigote o compund eterozigoti per mutazione
-
KIAA1985 proteina
-
Espressione: Nervose tessuti, includono periferici nervo
-
Contiene SH3 e TPR motivi
-
Funzione: Incerta, possibili ruolo in assemblaggio della proteina complessi
-
Clinica
-
Insorgenza
-
Età: Infantile (1 a 5 anni) o adolescenza
-
Precoce scoliosi: NCV può essere relativamente preservati
-
Altro pazienti con infantile neuropatia: Precoce perdita di
ambulation e respiratorie problemi
-
Scheletro: Main presentazione segni e disabilitante caratteristica
-
Scoliosi: Progressive; Insorgenza 3 a 9 anni; Chirurgia nell'adolescenza
-
Pes cavus: comune
-
Debolezza
-
Ritardato camminanti: Normale a 30 mesi
-
Distale: 100%
-
Prossimale: Tarda; Alcuni pazienti
-
gambe > braccia
-
Lentamente progressive
-
Atrofia: In regioni di debolezza
-
Perdita sensoria
-
Distale
-
Cutaneous perdita: Precoce in malattie decorso
-
Vibrazioni e articolazioni posizione perdita: Successivamente
-
Ariflessia nella maggior parte delle
-
No tremore o cerebellare Atassia
-
Prognosi
-
Molto lenta progressione della neuropatia
-
Lieve andatura malattie dopo 15 anni malattie durata
-
Qualche paziente in carrozzina in 20's to 40's
-
Laboratorio
-
CSF: Proteina probabilmente normale
-
Nervi Conduzioni
-
Velocità: Lenta; 10 a 34 M/s
-
Latenza distale: Prolungata
-
Potenziali sensori: Ridotta o assenza
-
Patologia: Neuropatia demielinizzante
-
Grave deplezione di grossi mielinizzati assoni
-
Mielina: Sottili e grossi focale Spessaenings
-
cellule di Schwann
-
Citoplasmica extensions, specialmente dal attorno non mielinizzati
assoni
-
Surrounded by multipla basale membrane
-
Bulbi a cipolla: Relatively sottili vs. CMT1A
-
Occasionale tomacula
CMT 4D: HMSN (Demielinizzanti) e perdita di udito (Lom tipo)
l N-myc
Downstream-Regolati Gene 1 (NDRG1)
; Cromosoma 8q24; Recessive; ?
correlazione to HMSN 4A
-
Mutazione: Premature-codone di terminazione una posizione 148
-
Proteina
-
Espressione: Ubiquitous; Alte livelli in cellule di Schwann
-
Possibile funzioni
-
Crescita arresto e Cell differenziazione
-
Signaling proteina shuttling fra citoplasma e the nucleo
-
Epidemiologia: Gypsies in Lom, Bulgaria, Slovenia, Spain e Italia
-
Decorso della malattia
-
Insorgenza: 1 a 10 anni di età; Disordinata andatura; Deformità nei piedi
-
Progressione: Grave disabilità by 5o decade
-
Clinica
-
Motorio: Distale > prossimale; Lingua atrofia; Mani debolezza @ 5 a 20
anni
-
Perdita sensoria: Tutte Modalità ; Distale
-
Riflessi tendinei: Assenza
-
Scheletro: Pes cavus (50%); Scoliosi (20%)
-
Sordità: Insorgenza 3a decade; Parlata percezione ridotta; Il nervo uditivo lesion
-
Vestibular disfunzione: Asintomatica
-
Intelligenza: Normale
-
Portatori: Asintomatica; Normale NCV
-
Elettrofisiologia
-
NCV: Molto lenta (9 to 20 M/s); Prolungata terminale latenza
-
Perdita assonale: Assenza o ridotta sensorio potenziali
-
Hearing: Lenta BAEP (Demielinizzazione in VIII nervo); Sensorioneurale tipo
perdita di udito
-
EMG: Denervazione in distale gambe
-
Patologia
-
Demielinizzazione
-
Bulbi a cipolla
-
Specialmente giovanier pazienti
-
La regressione con l'aumento dell'età
-
Older pazienti: cellule di Schwann processi in circular Modello;
Poche bulbi a cipolla
-
Mielina non compattata
-
cellule di Schwann inclusioni: adassonale citoplasma; Pleomorphic
-
Perdita assonale: Grave perdita di Assoni mielinizzati; Distale > prossimale
-
Assonale inclusioni: Curvilinear corpi; Simile to patologia in
vitamina E carenza
-
Endoneuriale collagenization: Vasi surrounded by multipla layers
di lamina basale
CMT 4F3
l Periassina
; Cromosoma 19q13.13-q13.2;
Recessive
-
Mutazioni del gene: Nonsenso e frameshift
-
Periassina proteina
-
Membrane proteina
-
3 Localizzazioni durante sviluppo
-
Nucleare: Precoce
-
adassonale: Membrana plasmatica
-
Abassonale: Con mielina maturazione; Schmidt-Lanterman incisure;
Paranodal membrane
-
Funzione
-
Interagisce con distrofina-correlate proteina 2 (DRP2): Via PDZ
domini
-
FormUn insieme a cellule di membrana di Schwann con distroglicano
-
? Participates nella membrana-proteina interazioni che stabilizzare
fogli mielinici
-
? Interagisce cnellamina basale circostante cellule di Schwann
-
isoformae contengono PDZ motivo e Nucleare localizzazione segnali
-
Epidemiologia: Numerose ethnicities
-
Libanese Shiite Muslim; North ispanoamenricana; nord europee;
Vietnamese
-
Clinica: Dejerine-Sottas
-
Insorgenza: Prima infanzia; 4 settimane a < 7 anni
-
Precoce sviluppo
-
Gestazione e Delivery: Normale
-
Ritardo motorio: Sitting età 12 a 18 mesi; Camminare e Talking a 2
anni
-
Andatura: Larga basate; Ataxic
-
Debolezza
-
Grave
-
Distale gambe a 9 to 10 anni
-
Mani a 14 a 15 anni
-
Progressione: Lenta
-
Perdita sensoria
-
più grave che altre DSS o CMT
-
Atassia (80%)
-
Pansensorio
-
braccia e gambe
-
Nervi craniali: Normale
-
Riflessi tendinei: Assenza
-
Scheletro: Lieve cifoscoliosi; Pes cavus (75%)
-
Clinica: CMT fenotipo con Early perdita Sensoria
-
Genetica: C715X mutazione, omozigote
-
Insorgenza: Infantile; Disturbi dell'andatura
-
Caratteristiche cliniche
-
Scoliosi: Grave a 10 anni
-
Debolezza: Distale; gambe e braccia; Simmetrica
-
Perdita sensoria: Grave; Distale; Tutte Modalità
-
Riflessi tendinei: Assenza
-
Progressione: Molto lenta
-
Laboratorio
-
Studi di conduzione nervosa
-
Motorio: Assenza motor potenziali o Gravemente abbassata Velocità
-
Potenziali sensori
-
Solita: Assenza
-
Possono essere preservati nella fanciullezza, specialmente più leggera sindromi
-
Patologia dei nervi: Perdita assonale (Grave); Bulbi a cipolla; Ipomielinizzazione
CMT 4H52
l Cromosoma
12p11.21-q13.11; Recessive
-
Epidemiologia: Libanese e algerine famiglie
-
Genetica: Simile locus to CMT 2G
-
Clinica: Grave
-
Insorgenza
-
Età: 10 a 24 mesi
-
Ritardato camminanti: 15 a 36 mesi
-
Andatura: Uncostante
-
Motorio: Debolezza distale Degenerazione; Piedi > Mani
-
Riflessi tendinei profondi: Assenza
-
Perdita sensoria: Lieve, Symmetrical; gambe > braccia
-
Scheletro: Scoliosi; Pes equinus
-
Decorso: Lenta progressione
-
Laboratorio
-
Elettrofisiologia
-
SNAPs: Assenza
-
NCV: Lenta; Latenza distale prolungato
-
CMAPs: Basso ampiezza
-
Patologia
-
Perdita assonale
-
Ipomielinizzazione
-
Bulbi a cipolla: Sottili
HMSN-Russe (HMSNR)5
l Cromosoma 10q23.2;
Recessive
-
Epidemiologia
-
Northern Bulgarian Famiglia (Russe)
-
Romanian e spagnoli Gypsies: Simile aplotipo to Bulgarian Famiglia
-
Insorgenza
-
Età: 8 a 16 anni; Media 11 anni
-
Debolezza nelle gambe
-
Clinica
-
Debolezza
-
Distale: Anteriori e Posteriori gambe
-
Simmetrica
-
Distale gambe: Insorgenza a 8 a 16 anni
-
Mani: Insorgenza 10 a 43 anni; Media 22 anni
-
Progressione
-
Continuo
-
Grave debolezza
-
Distale to knees e elbows by 4a to 5o decadi
-
Prossimale gambe
-
Perdita sensoria: Distale; Mani e piedi; Panmodale; Pronunciato
-
Riflessi tendinei: Assenza
-
DeFormities
-
piedi: Pes cavus; Clawed pollici; 100%
-
Mani: Clawing; 80%
-
Neuropatiche giunture: Occasionale
-
Craniale nervo coinvolgimento: Occasionale ptosi, debolezza facciale o
disfonia
-
Laboratorio
-
NCV: Perdita assonale
-
SNAPs e distale CMAPs assenti
-
NCV: moderatamente ridotta; 30 a 35 M/s
-
Aumentati soglia per electical stimolazione dei nervi
-
Patologia dei nervi
-
Perdita assonale: Specialmente grossi mielinizzati assoni
-
Rigenerazione
-
Ipomielinizzazione: Uniformemente ridotta Spessaness di fogli mielinici
HMSN con CNS o Craniale nervo coinvolgimento
HMSN con CNS o Craniale nervo: Dominante, Demielinizzanti
HMSN con CNS o Craniale nervo: Dominante, Assonale
HMSN con CNS o Craniale nervo: Recessive, Neuropatia assonale
-
HMSN e Agenesi di Corpus Callosum: Andermann Sindrome:
l KCC3 (SLC12A6)
; Cromosoma 15q13-q15;
Recessive
-
Genetica
-
Mutazione tipo: Proteina troncazioni
-
francese-Canadian mutazione: 2436delG
-
Mutazioni impair funzione di cotrasportoer
-
Knockout topo sviluppo sindrome simile
-
Periferici nervi hanno hypomielinizzazione
-
Normale Corpo calloso: Altro fattori può essere coinvolta in
Agenesi
-
Simile locus to
SPG (Murillo)
-
KCC3 proteina
-
Potassio-Cloro
cotrasportoer
-
Localizzazione
-
Cervello e Midollo spinale
-
cellule
-
CNPase positive oligodendroglia in materia bianca
-
Basolateral membrane del coroide plessi epithelial cellule
-
Aumentati attività con cellule gonfi
-
Famiglia
-
Diuretic sensibile catione-chloride cotrasportoers
-
Altro membri
-
Tiazina sensibile Na+-Cl-
cotrasportoer (NCC)
-
Na+-K+-2Cl-
cotrasportoers (NKCC 1 e 2)
-
Glicoproteine: Alte mannosio; Possono essere complesso
-
Epidemiologia
-
francese-Canadian: Charlevoix-Saguenay geograficaal focus; 1 in
2,100 nati vivi
-
KCC3 mutazioni quindi trovata in non-francese-Canadian famiglie
-
Caratteristiche cliniche
-
Nervi craniali: Simmetrica o Asimmetrica coinvolgimento
-
Ptosi
-
Debolezza facciale
-
Oftalmoplegia: Ridotta upgaze; Altro
-
Neur(on)opathy
-
Motorio
-
Precoce: Ipotonia
-
Grave debolezza: Distale e prossimale
-
Progressive
-
Raramente camminano indipendentemente
-
Perdita sensoria
-
Ariflessia
-
Tremore
-
CNS
-
Cognitive
-
Ritardo mentale
-
Atipica psychosis: Insorgenza nell'adolescenza; Episodica
-
Attacchi epilettici
-
Atrofia ottica
-
caratteristiche dismorfiche
-
Faccia: lunga; Ipertelorismo
-
Brachicefalia; Alte palato arcuato
-
Toes: Sindattilia di 2a e 3a; Overiding 1a
-
Scoliosi
-
Polmonare restrizione
-
Laboratorio
-
Elettrodiagnostica: Perdita assonale
-
Assenza sensorio potenziali
-
Lievemente ridotta motor NCV
-
CSF: Proteina lievemente aumentato
-
Patologia
-
Agenesi del corpo calloso
-
Parziale o complete (58%)
-
Dovuta un difetto in assoni migrazione attraverso linea mediana
-
Nervi craniali: Rigonfiamenti assonali con pochi neurofilamenti
-
Nervi periferici
-
grossi mielinizzati assoni: Ridotta
-
Rigonfiamenti assonali con pochi neurofilamenti
-
HMSN, neuropatia ottica ± perdita di udito
l Recessive
e collegata al cromosoma X Forma
-
Paraplegia spastica quindi riportate in 1 Famiglia
-
HMSN e sordità
l Autosomica
recessiva
-
Clinica
-
Polineuropatia
-
Decorso: Insorgenza precoce; Lentamente progressive
-
Debolezza: Distale; braccia e gambe; Simmetrica
-
Sensoria: Ridotta proprioception nelle gambe; + Romberg
-
Sordità sensorioneurale: Congenita
-
Ritardo mentale: Lieve
-
± Segni piramidali; Atrofia cerebellare
-
Riflessi tendinei: Assenza
-
Nervi conduzione
-
Lenta motor Velocità: Median < ulnar
-
Assenza sensorio potenziali
-
Patologia
-
Vedi anche: CMT 2E;
Perdita di udito
-
Vedi anche:
Neuropatie ad insorgenza infantile
HMSN con CNS o Craniale nervo: Recessive, Demielinizzanti
-
HMSN (Demielinizzanti) e perdita di udito (Lom tipo; CMT 4D)
-
Leucodistrofia metacromatica
l Arylsulfatase A
 ; Cromosoma 22q13.31;
Recessive
; Cromosoma 22q13.31;
Recessive
-
Caratteristiche cliniche
-
CNS: Ritardo mentale; Ottiche Atrofia; spasticità
-
Polineuropatia
-
Giovanile insorgenza Forma: Neuropatia può essere presentazione caratteristica
-
Debolezza: Prossimale e Distale
-
Ipotonia
-
Riflessi tendinei: Diminuita o assenti
-
Perdita sensoria: Lieve; Distale
-
Trattamento: Corticosteroidi Possono produrre sintomatiche benetfit
per 2 a 3 anni
-
Laboratorio
-
NCV: Demielinizzazione
-
Rallentato: Variabili Early; Più uniforme con progressive
malattie
-
Prolungata F-onde
-
Non blocco di conduzione
-
Nervi Patologia
-
Ipomielinizzazione
-
Metacromatici inclusioni in cellule di Schwann e
macrofagi
-
Lamellar o scuro ultrastrutture
-
CSF proteina: Alte
-
Escludono la:
-
multipla Sulfatase Carenza (Recessive)
-
Activator Proteina (Prosaposin) Carenza
l
Cromosoma 10q21-q22; Recessive
-
Galactosylceramide Lipoidosis (Krabbe)
l
Galactosylceramide β-galattossidasi
; Cromosoma 14q31; Recessive
-
fanciullezza38
-
CNS
-
Ritardo mentale
-
Irritabilità: Pianto eccessivo
-
Ottiche Atrofia
-
Tone: spasticità; Ipertonia (Occasionale ipotonia)
-
Polineuropatia
-
Insorgenza: < 6 mesi
-
Clinica
-
Possono essere presentazione sindrome (Fino ai 25% dei pazienti):
Prima CNS
-
Ritardo motorio
-
Ipotonia
-
Ridotta movimenti
-
Riflessi tendinei: Ridotta o assenza
-
Altro CNS caratteristiche sviluppo nel corso di mesi
-
Elettrofisiologia: Neuropatia demielinizzante
-
Velocità di conduzione nervosa: Lenta
-
Latenza distale: Prolungata
-
Blocco di conduzione: Occasionale
-
F-onde risposta: Ritardato o assenza
-
CMAP: Lievemente ridotta ampiezza
-
Biopsia dei nervi
-
Ultrastrutture: Curved tubulare inclusioni in cellule di Schwann citoplasma
-
Luce microscopia: Maggio mostrare pochi caratteristiche della mielina
patologia
-
Successivamente-insorgenza Forma29
-
Insorgenza
-
Età: > 10 anni; Gamma 13 a 47
-
debolezza degli arti
-
Disturbi dell'andatura
-
Midollo spinale
-
spasticità: gambe e Vescica
-
Riflessi tendinei: Vivaci o assenza
-
Polineuropatia: Predominantemente motor
-
Possono essere asimmetrico
-
Debolezza
-
Pes cavus
-
Fascicolazioni: Muscoli prossimali
-
Decorso: Progressive nel corso di anni
-
Altro CNS segni
-
Disartria
-
Atrofia ottica
-
Atassia
-
Demenza
-
Genetica
-
La maggior parte mutazioni in regioni codificante per 50 kDa subunità
-
Occasionale mutazioni in 30 kDa subunità
-
Laboratorio
-
CSF: Proteine alte
-
Elettrofisiologia: Neuropatia demielinizzante; NVC lenta
-
MRI: Tratto corticospinale e periventricolare demielinizzazione
-
Patologia
-
Mielina
-
Ipomielinizzazione degli assoni: Uniformemente thnella fogli mielinici nella maggior parte degli assoni
-
Alcuni pazienti con demielinizzazione e bulbi a cipolla
-
Inclusioni
-
Localizzazioni: cellule di Schwann; Histiocytes
-
Struttura
-
Sagoma: diritta o Leggermente curved, Crystalloid e
Prismatic
-
Contenente: Chiari e empty on paraffin e ultrastrutture
-
Perdita assonale
-
Glicoproteina carente di carboidrato sindrome
Refsum Sindromi
-
Refsum malattie: ad insorgenza infantile (< 20 anni)
: Classic Form
l fitanolo-CoA
Hydroxylase
; Cromosoma 10pter-p11.2;
Recessive
-
PHYH mutazioni genetiche
-
Missenso il più comune
-
La maggior parte mutazioni inactivate funzione enzimatica
-
Alcune mutazioni produce proteina con normale attività, ma
difettose targeting to perossisomi
-
fitanolo-CoA Hydroxylase
-
Enzima funzione: Converte phytanoyl-CoA to
2-hydroxyphytanoyl-CoA
-
fitanolo-CoA + 2-ossoglutarato + O2 =
2-hydroxyphytanoyl-CoA + succinato + CO2
-
1a passo nel α-ossidazione di acido fitanico
-
localizzazione subcellulare:
Perossisomi
-
Localizzazione cellulare: fegato, reni, e Cellule T
-
Deficiente nel fegato nella sindrome di Zellweger
-
Insorgenza
-
Night CECITA'
-
5 a 20 anni di età
-
Neuropatia demielinizzante
-
Distribuzione di segni: Simmetrica; Distale progressione to Prossimale
-
Debolezza: Distale; gambe > braccia
-
Perdita sensoria: Vibrazioni e Propriocezione > Dolore e Temperatura
-
Riflessi tendinei: Ridotta o assenza
-
Nervi dimensione: Ingranditi in alcuni pazienti
-
Decorso
-
Maggio presenti subacutely over settimane
-
Progressive o Recidevante
-
Nervi conduzione : Molto lenta motorio e sensorio Velocità (Alcune < 10
M/s)
-
CSF Proteina: Molto Alte
-
Patologia dei nervi
-
Ipertrofia: Prossimale > distale
-
Bulbi a cipolla: grossi
-
Assoni mielinizzati: Ridotta numeri
-
Ultrastrutture: Inclusioni in cellule di Schwann citoplasma
-
Nervi craniali
-
CNS
-
Clinica: Atassia
-
Patologia: Ridotta numeri delle cellule di Purkinje e Neuroni in
oliva inferiore, Dentate, Vestibular, Coclear e Rossa nuclei
-
retinite pigmentosa
-
granulare
-
Concentric costrizione di Visione campi
-
Ridotta ERG
-
Sistemico
-
Cardiaco Insufficienza: Può causare la morte improvvisa; Insorgenza dopo fanciullezza
-
Ichthyosis
-
Il diabete
-
Scheletro: Corta 4a metatarsal; Epiphyseal displasia; Sindattilia
-
Biochimica
-
elevata serico acido fitanico (Branched catena acidi grassi)
-
Ridotta ossidazione di acido fitanico nei fibroblasti
-
Perosossisomi disfunzione
-
Trattamento: Basso acido fitanico dieta
-
Refsum malattie: Adolescent o giovani adulti
(Tipica Form)
l PEX7
; Cromosoma 6q22-q24;
Recessive
-
PEX7 mutaziones35
-
Refsum malattie
-
Composizione eterozigoti: 1 Mutazione con lieve effetto in ogni
paziente
-
Y40X; 12-18dupGTGCGGT Frameshift; T14P
-
Mutazioni causano anche: rizomelica Condrodisplasia puntata tipo
1
-
Proteina: Perossina 7
-
Funzione: Import nella
perossisoma di proteine contenente perossisomici segnali bersaglio
tipo 2 (PTS2)
-
Insorgenza: 2a to 4a decade
-
Clinica
-
Atassia
-
Polineuropatia
-
Perdita sensoria
-
Debolezza
-
Ingrandimento dei nervi
-
Anosmia
-
Retinite pigmentosa
-
Scheletro: Corta 5o metacarpal; Pes cavus
-
Laboratorio
-
Plasmalogen sintesi: Deficiente
-
Refsum's: ad insorgenza infantile
l Perossina-1 (PEX1)
; Cromosoma 7q21-q22;
Recessive
-
Proteina
-
Richiesta per perossisomica matrice importare le proteine
-
Genetica
-
mutazione omozigote (Gly843Asp) trovata in
-
Heterozygosity per questa mutazione (? altre mutazione) trovata in
-
Zellweger's; Neonatale Adrenale leucodistrofia; Infantile
Refsum's
-
Clinica
-
Ritardo mentale
-
Neuropatia:
Demielinizzanti
-
Sordità
-
Retinite pigmentosa
-
GI: Epatomegalia; Steatorrhea
-
Scheletro: Dismorfismo facciale; Crescita insufficiente; Osteopenia
-
Biochimica
-
Hypocolesteroloemia
-
Accumulazione di acido fitanico, pipecolic acido, molto lunghezza catena
acidi grassi
-
Patologia
-
CNS: leucodistrofia
-
Perossisomi: Ridotta o assenza
-
Insorgenza adulta Refsum's con pipecolicacidemia
l ? Perosossisomi
malattie; Cromosoma 10p; Recessive

|
HMSN con CNS o Craniale nervo: collegata al cromosoma X, Demielinizzanti
HMSN con segni piramidali e materia bianca cerebrale lesions40
l Semi-Dominante
-
Epidemiologia
-
giapponese Famiglia: 3 gravi maschi, 2 lieve femmine
-
Genetica: No specifico X-collegamento definiti
-
Insorgenza
-
Età: Adulto, 3a decade
-
Sintomi: Disturbi dell'andatura
-
Clinica
-
Debolezza: Distale; Mani e piedi
-
Degenerazione: Distale; Mani e piedi
-
superiori neuroni motori segni
-
spasticità: gambe > braccia
-
Plantar responces: Estensore
-
Riflessi tendinei
-
Ankles: Assenza
-
Altro: Vivaci
-
Perdita sensoria: Distale; Panmodale
-
Nervi craniali: Normale
-
Laboratorio
-
Elettrofisiologia
-
NCV: 13 a 36 M/s
-
CMAPs: Assenza o ridotta ampiezza
-
SNAP ampiezza: Ridotta o assenti
-
Evocate potenziali: anormale centrale somatosensory e motor
percorsi
-
Biopsia dei nervi: Non-diagnostico
-
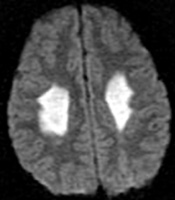
MRI: Hyperintense lesioni inperiventricular materia bianca
-
Femmine eterozigote: Più lieve della maschi
-
Insorgenza: Adulto
-
Clinica
-
Degenerazione e debolezza
-
superiori neuroni motori
-
Andatura: Lievemente spastica
-
Plantar riflessi: Estensore
-
Riflessi tendinei
-
Ankles: Assenza
-
Altro: Vivaci
-
Elettrodiagnostica
Ereditarie Neuropathies - Altro
α-metilacil-CoA racemasi (AMACR) carenza
l AMACR
; Cromosoma 5p13.2-q11.1;
Recessive
-
Enzima funzione
-
Localizzazione:
Perosossisomi
-
Metaboliche:
β-ossidazione percorso di una catena ramificata acidi grassi
-
Mutazioni puntiformi produce inattivo proteine
-
Epidemiologia: 4 pazienti sporadici identificate
-
Insorgenza
-
Infantile to Precoce adulti
-
Encefalopatia
-
Clinical24
-
Polineuropatia
-
Insorgenza adulta: Neuropatia presenti in 2 pazienti adulti ma probabilmente
non i bambini
-
Debolezza: gambe > braccia
-
Perdita sensoria
-
Elettrofisiologia
-
Perdita assonale in 2 pazienti; Demielinizzazione in un altro
-
Sensoria e Motorio coinvolgimento
-
CNS: Variabilmente presenti
-
Paraparesi spastica
-
Ritardo nello sviluppo (Lieve): Difficoltà ad apprendere
-
Attacchi epilettici
-
Encefalopatia: Episodica
-
Retinite pigmentosa
-
Laboratorio
-
Pristanic acido (a ramificata catena acidi grassi) in plasma: Aumentati
-
C27-bile-acido intermedi in plasma: Ridotta
-
AMACR attività assenti nei fibroblasti
HMSN con
formazione di minifascicoli e 46XY Pure Gonadal Dysgenesis25
l Sporadica vs. Recessive
-
Epidemiologia: Singola giapponese paziente
-
Genetica: Normale SRY e DHH geni
-
Insorgenza: 39 anni; Distale intorpidimento e debolezza
-
Clinica
-
Neuropatia
-
Debolezza: Distale; Moderate; braccia e gambe
-
Sensoria: Distale; Panmodale; Grave
-
Riflessi tendinei: Assenza
-
Gynecological
-
Breasts: Poorly sviluppato
-
Pubic capelli: Sparse
-
Vagina: Blinded
-
Uterus: Immature
-
Gonads: Streak
-
CNS: Normale
-
Laboratorio
-
Hormones: LH e FSH alto; Testosterone e Estradiol basso
-
Elettrofisiologia
-
SNAPs: Assenza
-
Motorio: CMAP sottili; moderatamente abbassata NCV
-
EMG: Denervazione
-
Patologia dei nervi
-
Minifascicoli: 60 sottili fascicles in sural nervo
-
Assonale densità: Ridotta sottili e grossi mielinizzati fibre; Normale
non mielinizzati assoni
-
fogli mielinici: Sottile
-
Perineurial cellule: anormale lamina basale
-
Vedi anche:
Polineuropatia con Minifascicoli, 46,XY disgenesi gonadiale e ritardo mentale
HMSN con
Congenita vertical talus49
l HOXD10
; Cromosoma 2q31; Dominante
-
Epidemiologia: Singola Northern Nuove York bianchi Famiglia; italiane discendenti
-
Genetica: Mutazione missenso; M319K
-
Clinica
-
Congenita vertical talus (CVT; Rocker-bottom foot)
-
Rigid dorsale dislocation di navicular over collo di talus
-
Bilaterale o Unilaterale
-
Neuropatia
-
Insorgenza precoce
-
Lieve
-
Piedi solo
-
Possono essere asimmetrico
-
Laboratorio: Non disponibile
Vedi anche:
Neuropatie ad insorgenza infantile
 paziente informazioni
paziente informazioni
Support gruppi
Ritorno a
Indice delle polineuropatie
Ritorno a
Neuromuscolare home page
Riferimenti
1. Am J Hum Genet 1999;65:722-727, Malattie neuromuscolari 2003;13:60–67
2. Genomics 1999;62:344-349, Esseri umani molecolare Genetica 2003;12:349–356
3. Am J Hum Genet 2001;68:325-333
4. Am J Hum Genet 2000;67, Cervello 2007;130:394–403
5. Am J Hum Genet 2000;67:664-671, Ann Neurol 2001; Online Aug 7
6. Neurologia 2000;55:392-397, Am J Hum Genet 2003;72 Online April3
7. Malattie neuromuscolari 2000;10:497-502
8. J Neurol 1999;246:107-112
9. JNNP 2000;69:806-811, Cervello 2003;126:134-151
10. JNNP 2000;69:799-805
11. Neurologia 2000;55:1552-1557
12. Am J Hum Genet 2001;68:269-274, Malattie neuromuscolari 2004;14:301–306
13. Neurologia 2001;56:100-103
14. Malattie neuromuscolari 2001;11:27-34
15. Lancet 2001;357:267-272
16.
Hum Mol Genet 2001;10:947-952
17. Neurologia 2001;56:A315, Malattie neuromuscolari 1998;8:392–393, Am. J. Hum.
Genet. 2001;69:000–000
18. Malattie neuromuscolari 2001;11:400-403
19. Eur J Hum Genet 2001;9:646-650
20. Am J Hum Genet 2002;70:244-250, Neurologia 2003;60:22-26
21. Neurologia 2001;57:1906-1908
22. Nature Genet 2002;30:21-22, Neurologia 2002;59:1865–1872
23. Nature Genet 2002;30:22-24, Arch Neurol 2003;60:598-604, Cervello 2003;126:
Agosto
24. JNNP 2002;72:396-399
25. Ann Neurol 2002;51:385–388
26. Am J Hum Genet 2002; On-Line April
27. Malattie neuromuscolari 2002;12:399–404, J Neurol Neurosurg Psychiatry
2002;73:762–765, Neurologia 2003;60:1151–1156
28. Neurologia 2002;58:1769–1773
29. Malattie neuromuscolari 2002;12:386-391
30. Ann Neurol 2002;52:429–434
31. J Neurol 2002;249:1298-1302
32. Muscoli Nervi 2002;26:608–621
33. J Cell Sci 2002;115:4937-4946
34. Muscoli Nervi 2003; Online Dicembre 2002
35. Am J Hum Genet 2003; On-Line gennaio
36. J Neurol 2002;249:1629-1650
37. Neurologia 2003;60:506–508
38. Pediatr Neurol 2003;28:115-118
39. Cervello 2003;126:590-597
40. Muscoli Nervi 2003;28: 623–625
41. Am J Hum Genet 2003;73:1106–1119
42. Neurologia 2003;61:1154-1155
43. Am J Hum Genet 2003; Online Novembre
44. Muscoli Nervi 2004;29:205-210, Acta Neurol Scand 2003;108:352-358,
Neuromuscul Disord. 2003;13:827-829
45. Neurologia 2003;61:1457-1458
46. J Med Genet 2004;41:193–197
47. Esseri umani Genet 2004;114:527–533, Esseri umani Genet 2004; Online Novembre
48. J Peripher Nerv Syst. 2004;9:124
49. Am J Hum Genet 2004; Online Maggio
50. J Neuropath Exp Neurol 2004;63:1167-1172
51. J Neurol 2004 Dec;251(12):1491-1497
52. Hum Mol Genet 2005; Online March
53.
Ann Neurol 2005;57:749–754
54.
Neurologia 2005;64:1964–1967
55.
J Cell Sci 2005 Jun 28,
Hum Mol Genet 2005;14:1405-1415
56.
JNNP 2005;76:1109-1114
57.
Neurologia. 2005;65:197-204,
Neurologia. 2005;65:496-497
58.
Neuromuscul Disord 2005 On line Sep 27
59. Nature Genetica 2005; Online Jan 22
60.
Ann Neurol 2005;59:276–281
61. J Neurol Sci 2005; Online
62.
JNNP 2006;77:963-966,
J Comp Neurol 2006;498:252-265,
Neuromuscul Disord 2006;16:308-310
63.
Neurologia 2006;67;2016-2021
64.
Neurologia 2006;67;2250-2252
65.
Ann Neurol 2006;59:358–364
66.
Otol Neurotol 2005;26:405-414
Illustration by J. Kwon, MD

{kind=link}