 Traduzione di Natale
Marzari
Traduzione di Natale
Marzari
Home,
Ricerca,
Indice alfabetico,
Collegamenti,
Patologia,
Molecole,
Sindromi,
Muscoli,
Giunzioni neuromuscolari,
Nervi,
Spinale,
Atassia,
Anticorpi e Biopsia,
Informazioni per i
pazienti
|
 |
Traduzione del
13
gennaio 2011
SLA SPORADICA
SLA sporadica: Forma comune
- SLA: Epidemiologia 7
- Statistiche sulla frequenza
- Incidenza/100.000/anno
- Generale: 0,4 ÷ 2; Maschi = 2,1; Femmine 1,8
- 65 ÷ 74 anni: Maschi = 10,2; Femmine = 7,4
- Aumento negli anni '90 in: Femmine; Pazienti > 75 anni;
Europa del sud
- Prevalenza: 3 ÷ 8 per 100.000
- Il rischio aumenta con l'età fino ai 74 anni
- Rischio di SLA per l'intera vita (Vivendo per 70 anni)
- Generale: 1:350 ÷ 1:2000
- Sesso: Femmine 1:450; Maschi 1:350
- Mortalità
- Causa 1 ogni 700 morti
- Tasso: 1,9/100.000/anno
- Geografia: Distribuzione in tutto il mondo
- Frequenza generale: Uniforme
- Eccezioni: Varianti di Guam (Chamorro) e della penisola Kii
del
Giappone
- Maschi:Femmine
- Paesi occidentali 1,5:1
- India 3:1
- Pazienti con insorgenza più giovanile: Maggiore predominanza maschile
- Frequenza della SLA famigliare vs le atre SLA: 5%
36
- Età dell'insorgenza
- Media: 46 ÷ 63 anni
- USA ed Europa: 56 ÷ 63 anni
- India: 46 anni
- Prima dei 20 anni: Solo casi rari
- Genetica
- Allele MAO-B5
 2:
Più tarda con allele (60 anni); Più precoce con altri (52 anni)
2:
Più tarda con allele (60 anni); Più precoce con altri (52 anni)
- Difetto genetico con CNTF omozigotico
 8
8
- Presente nel 2% dei normali
- Insorgenza più precoce di SOD e SLA sporadiche: 48 vs 58 anni; Normale
la durata della malattia
- Associato con la perdita 15%
÷ 20% dei neuroni postnatale
nei topi
- Associazione con numerose varianti nel
gene SMN2
- SOD1:delezione di 50 bp nella regione del promotore
30
- Età dell'insorgenza della SLA: Aumentata
- Attività del SOD1: Ridotta
- Legame a SP1: Ridotto
- Associazioni della SLA: Malattia e gene
10
SLA: Possibile suscettibilità
e geni modificatori 26 |
| Gene |
Locus |
Variante |
| ApoE ε4
|
19q13 |
Genotipo ε4 |
| CNTF
|
11q12 |
Allele nullo |
| EAAT2
|
11p13 |
Bassa espressione |
| GluR2
|
4q32 |
Δ editante RNA |
|
NEFH
|
22q12 |
Delezioni KSP |
| SMN1
|
5q12 |
Numero di copie |
|
SMN2
|
5q12 |
Numero di copie |
|
VEGF
|
6p12 |
Promotore di SNP |
|
Dinactina
|
2p13 |
Mutazioni |
| FLJ10986
|
1 |
Linkage |
| ITPR2
|
12p11 |
Linkage |
| DPP6
|
7 |
Linkage |
| Progranulina
|
17q21.3 |
Mutazioni |
|
FIG4
|
6q21 |
Mutazioni |
CHRNA3
,
CHRNA4
CHRNB4
|
15q25,
20q13,
15q24 |
Mutazioni 33 |
| UNC13A
|
19p13.3 |
Collegamento all'Introne 21 34 |
PON1
PON2
PON3
|
7q21.3 |
Linkage |
| |
9p21.2 |
Linkage |
|
Atassina-2
|
12q24.1 |
Espansioni poliQ
Lieve: Glutamina 27–33 |
- Habitus: Magrezza per tutta la vita (2,2 x più alta)
- Storia di
- Sportività atletica universitaria: 1,7 x più alta
- Fumo 21
- Maggiore con: L'aumento del fumo
- Concorrenza del fumo nelle femmine: 1,7 x più alta
- Minore con l'aumentare del tempo dopo avere smesso
- ? Linfoma con la proteina M o con proteine elevate nel CSF
- Riarrangiamenti cromosomici 14
- Frequenza: 5,9% vs 0,05 ÷
0,1% nella popolazione generale
- Tipi: Traslocazioni bilanciate (3/85); Inversioni pericentriche (2/85)
- EAAT2 (trasportatore del glutamato)
:
Errori di processamento del RNA
- APOE
- Alti livelli plasmatici
- Correlati con rapido deterioramento e minore tempo di sopravvivenza
- Relative rischio = 0.647
- Fenotipo APOE: Correlazioni non chiare
-
Fattore di crescita vascolare endoteliale (VEGF)
18
- L'aplotipo omozigote produce poco VEGF
- Rischio di SLA 1,8 volte più alto
- Aplotipi: -2578A/-1,154A/-634G; -2578A/-1,154G/-634G
- Associato con bassi livelli di VEGF circolante
- Associazione trovata sia con la SLA sporadica che con la SLA famigliare
- Morte più precoce nei topi mutanti SOD con VEGF espressione
insufficiente
- Iperlipidemia 25
- Colesterolo totale o LDL alti: 2x più frequenti nella SLA che nei controlli
- Tasso LDL/HDL alto: 12 mesi sopravvivenza più a lungo nella SLA
- Progranulina (PGRN; GRN)31
- SLA
- Mutazioni SLA: Missenso; Regolatorio del 5'; Varianti nel DNA
- Associazioni cliniche alla SLA delle mutazioni IVS2 +
21G>A o IVS3-47-46insGTCA
- Malattia ad insorgenza più giovanile
- Sopravvivenza più breve
- Demenza frontotemporale: Mutazioni nulle
-
SMN1
- Aumentata frequenza (2x) di 1 o 3 copie del gene nella SLA
-
FIG4
32
- Mutazioni SLA
- Eterozigotiche
- Missenso o terminazione
- Frequenza: 1% ÷ 2% dei pazienti con la SLA
- Tipi di SLA
- Storia famigliare:
Famigliare o sporadica
- Modello clinico: SLA o
PLS;
Riflessi tendinei vivaci
- Vedi anche:
CMT 4J
- SLA: Caratteristiche cliniche
- Modello tipico: Segni nei neuroni motori superiori ed inferiori con sensazioni normali
- Debolezza
- Modelli di debolezza e di disfunzione: Caratteristiche comuni
- Debolezza degli arti
- Modelli iniziali
-
Asimmetrica
- Può coinvolgere la muscolatura prossimale o distale
- Modelli specifici
- Estremità superiori > inferiori
- Frequenza più alta nei pazienti di ascendenza africana
- Distale nelle estremità inferiori
- Più comune all'insorgenza della malattia
nella SLA ereditaria
-
Disfunzione bulbare 28

Dalla Bramwell:
Atlante di medicina clinica
SLA con
coinvolgimento bulbare
|
- Associata più frequentemente con disfunzione dei neuroni motori superiori
- Frequenza
- Più comune nelle femmine più anziane: 50% con presentazione bulbare
- Segni bulbari all'insorgenza: 20%
÷ 30% di tutti i
casi di SLA sporadica
- Caratteristiche
- Disartria
- Velocità del parlato: Lenta
- Modello del parlato
- Smozzicato
- Frasi corte
- Pause inappropriate
- Qualità della voce
- Ridotta
- Ipernasalità
- Gamma ridotta di accenti e suoni
- Disfagia
- Aspirazione
- Incapacità di saturare l'O2, o
- Aumentata frequenza respiratoria dopo
assunzione orale
- Tipi di cibo: Secco, Di dura consistenza o
sbriciolanti; Con poco liquido
- Tempo per il mangiare: Aumentato
- Saliva: Cattiva deglutizione; Sbavamento
- Labilità emozionale
- Associazione comune
- Affezione "Pseudobulbare"
- Altre caratteristiche bulbari
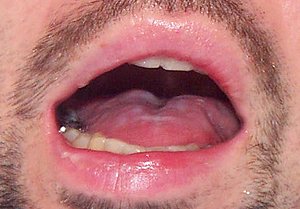
La lingua nella SLA: Debole con piccole
atrofie
e tendente alla protrusione a riposo
|
- Difficoltà a tossire volontariamente
- Laringospasmo (20%)
- Difficoltà ad usare dispositivi respiratori esterni
- Mascella
- Riflessi: Vivaci
- Difficoltà a chiuderla
- Lingua
- Movimenti: Lenti
- Massa: Spesso relativamente preservata per grado della disfunzione
- Insufficienza respiratoria
- Può aversi isolatamente
- Associazione frequente: Debolezza delle braccia
- Primi stadi spesso asintomatici
- Primo evento: Ipoventilazione
- Avviene dapprima nel sonno REM
- I muscoli accessori della respirazione diventano flaccidi
- Sintomi
- Dispnea
- Disturbi del sonno
- Cefalea mattutina
- Tosse inefficace: Debolezza dei muscoli espiratori
- Associazioni: Perdita del senso del gusto; Perdita di peso;
Depressione
- Dispositivo respiratorio esterno
- Più efficace in assenza di coinvolgimento bulbare
- Aree di debolezza con qualche specificità per la SLA
- Denervazione molto prossimale
- Debolezza della mascella: Chiusura; Apertura
- Voce
- Nasale, parlata incomprensibile
- Emissione continua di suono
- Lingua
- Debole
- Movimenti
- Lenti
- Di ampiezza ridotta
- Massa relativamente preservata: Spesso
- Segni associati
- Movimenti della mascella insieme alla lingua
- Riflessi della mascella: Iperattivi
-
Insufficienza respiratoria
Isolata
- Atrofia muscolare
- Spesso presente nel primo decorso della malattia
- Solitamente associata alla debolezza
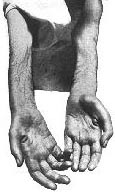
Atrofia delle mani e
delle braccia nella SLA
|
- Progressione: Regionale (Braccia, gambe o bulbare)
- I sintomi solitamente progrediscono dapprima nelle regioni
già affette
- La progressione continua una volta coinvolta la regione (bulbare,braccia, o gambe)
- Altre regioni coinvolte successivamente
- Si irradia solitamente alle regioni adiacenti
- Non sono prevedibili i tempi del coinvolgimento
- Si possono avere periodi di stabilizzazione sintomatica
- Rare le remissioni dei sintomi o dei segni
- Attività motorie spontanee
- Crampi
- Comuni nelle gambe, la notte
- Spesso si risolvono spontaneamente con la progressione della malattia
- Possono essere gravi successivamente nella malattia: Associati con spasticità
pronunciata
- Fascicolazioni
- La SLA presenta raramente fascicolazioni in assenza di debolezza
- Patogenesi: ? Maggiore persistenza della conduttanza Na+
negli assoni motori
- Neuroni motori superiori
- Modelli anatomici
- Bulbare: Causa la maggior parte della disfunzione della
lingua e bulbare
- Paraparesi
- Asimmetria: Comune negli arti, specialmente all'inizio della malattia
- Emiparetica corticale: Occasionale
- Riflessi tendinei
- Iperattivi (100%): Nelle aree con forza e debolezza normali
- Più comune il segno che rialza le dita dei piedi (30%
÷ 50%)
- Coinvolgimento bulbare associato alla labilità emozionale (75%)
- Può causare disabilità senza debolezza associata: Specialmente
nelle
gambe
- Correlazioni di laboratorio: Anormali
20
- Spettroscopia MR
- Stimolazione magnetica transcranica
- Dolore: Correlato all'immobilità
- Comportamento
- Alterazioni cognitive: Disfunzione dei lobi frontali (lieve) non insolita
- Affezione pseudobulbare: Riso e pianto non provocati; Comuni nel
coinvolgimento bulbare
- Demenza: 3% ÷ 5%
- Coinvolti raramente: Vescica; Intestini; Autonomo; Movimenti extraoculari;
Sensorio
- Cute: Immunoreattività del collagene di tipo IV ridotta
- Criteri diagnostici (con esami clinici, della patologia ed EMG)
- SLA definita: Malattia progressiva con
- Segni nei neuroni motori superiori ed inferiori in 2 regioni
spinali e bulbari,
o
- Segni nei neuroni motori superiori ed inferiori in 3 regioni spinali
- SLA probabile: Malattia progressiva con
- Segni nei neuroni motori superiori ed inferiori in 2 regioni e
- Segni nei neuroni motori superiori in una regione rostrale ai segni nei neuroni motori
inferiori
- Sindromi varianti
- Possibile diagnosi differenziale
-
Mielopatia o radicolopatia spondilitiche
- Varianti della malattia dei neuroni motori con segni solamente nei neuroni motori inferiori o superiori
-
Polimiosite
- Fascicolazioni
benigne
- Sclerosi multipla
- Malattia cerebrovascolare
- Sopravvivenza 29
- Media
- Sopravvivenza generale: 3 ÷ 4 anni
- Sopravvivenza dopo la diagnosi: ~2 anni
- Tempo dal sintomo iniziale alla diagnosi: ~1 anno
- Maggiore sopravvivenza 17
- Frequenza (> 10 anni): 4% ÷ 10%
- Caratteristiche all'insorgenza
- Più frequenti che nella popolazione SLA generale
- Età più giovane: Media 43 anni; 14 anni più giovani della media
- Segni dei motoneuroni superiori puri
- Nessuna differenza dalle popolazione SLA generale: Dal sesso;
Dal sito di
insorgenza
- Segni sia nei UMN che nei LMN all'insorgenza: 70%
- PLS:
Sopravvivenza più lunga
- Insorgenza più giovanile: Sopravvivenza più lunga
- Pazienti indiani 27
e africani: Sopravvivenza più lunga; Media 115 mesi
- Farmaci: Assunzione di calcio
- Prognosi più negativa per la sopravvivenza
- Clinica
- Età avanzante
- Pronunciata perdita di peso recente
- Breve tempo dall'insorgenza alla diagnosi
- Tasso rapido di perdita respiratoria e della forza nel corso
dei 6 mesi
dopo la diagnosi 9
- Insufficienza respiratoria
- Nessuna gastrostomia
- Farmaci: Aspirina; Acetaminofene
- Laboratorio
- Funzione polmonare: Scarsa; < 60% del previsto
- Siero
- Cloro: Basso; ? In relazione alla scarsa nutrizione
- Bicarbonato: Alto
- EMG
- CMAP bassi
- Decremento alla RNS
- SFEMG: Marcato tremolio; Bassa densità delle fibre
- ? Delezione omozigotica del
gene SMN2 5
- Più comune nella SLA sporadica
- Tempo di sopravvivenza: 2 anni più breve
- Morte
- Causa della morte: Insufficienza respiratoria la più comune
- Insorgenza di insufficienza respiratoria con coscienza ridotta a
causa dell'ipercapnia
- Morte: Solitamente tranquilla e avviene nel sonno
- Soffocamento insolita
- Caratteristiche di laboratorio
- EMG: Denervazione
- Spontanee attività
-
Fibrillazioni
-
Picchi d'onda positivi
-
Fascicolazioni 35
- Disturbo dei neuroni motori inferiori associato con
- Tasso di scatenamento delle fascicolazioni:
Aumentato
- Fascicolazioni doppie: Più comune
- Fascicolazioni complesse e instabili
- Si hanno sia nella SLA che nelle sindromi benigne
- SLA: Durata più breve e più accessi che nelle sindromi benigne
- Dimensioni delle unità motorie: Normali o grandi
- Distribuzione
- Prossimale e distale
- In numerose regioni del corpo
- Denervazione paraspinosa toracica
- Alcune specificità per malattie dei neuroni motori
- Si può avere anche nelle neuropatie motorie
- Siero
- CK: Normale ÷ 4x più alto
-
Angiogenina: Livello medio elevato del 20%, specialmente nella
SLA spinale 23
-
VEGF: Normale
- Lipidi: Colesterolo, LDL e trigliceridi lievemente ridotti con FVC
< 70%
- CSF: Solitamente normale
- MRI e imaging del cervello
- Alterazioni della corteccia
- Segnale T2 di intensità ridotta nella corteccia motoria (primaria e secondaria): Non specifico per
la SLA
- Acetilaspartato N ridotto alla MRI
- Può essere correlato con la spasticità: Tasso di
tambureggiamento con le dita
ridotto
- Cingolato anteriore e corteccia temporale anormali
- Anormalità nella materia bianca subcorticale
- Regioni del tratto corticospinale: Segnale T2 aumentato; Specialmente rostrale
- Probabilmente 2° alla perdita assonale (Degenerazione Walleriana)
- Corpo calloso
- Nessuna forte relazione al coinvolgimento clinico
dei neuroni motori superiori
- ? relazione ai movimenti a specchio
- Patologia
-
Cellule del corno anteriore
- Restringimento e perdita dei corpi cellulari
- Decorso della perdita dei corpi cellulari
22
- Inizialmente: Rapida
- Successivamente: Più lenta
- ? Associata con
Apoptosi
- Risparmiati: Nuclei di Onufrowicz, o Onuf
- Neuroni parasimpatici nella midollo spinale sacrale
- Innervazione dell'intestino e della vescica
- Aggregati nei neuroni motori spinali
- Contenenti SOD1:
SLA SOD1
- Contenenti
FUS e
TDP-43: SLA sporadica;
SLA ereditaria, eccetto la SLA SOD1
- Ubiquitina: Tutte le SLA
- Sferoidi (Neurofilamenti):
Negli assoni prossimali
- Corpi cellulari di Bunina (Inclusioni granulari dense): Nel
citoplasma dei corpi cellulari
- Corpi cellulari di Hirano
- Inclusioni a forma di bastoncelli con
filamenti paralleli ubiquitinati
- Maggiormente preminenti nei neuroni motori inferiori
- Altro sulla midollo spinale
- Patologia
- Tratti corticospinali: Perdita di assoni mielinizzati
- Colonna posteriore del midollo spinale: Risparmiata eccetto che nella
SLA famigliare
- Proliferazione astrogliale
- Infiammazione: Piccola o nessuna
- Inclusioni: Positive alla ubiquitina e TDP-43
24
- Presenti nel citoplasma dei neuroni e della glia della SLA sporadica
- Filamentose o rotonde
- Trovate anche nella: SLA con demenza; Non FALS SOD1
- Non presenti nella FALS SOD1
- Biochimica del midollo spinale 12
- Livelli aumentati di
- Ceramide C16:0, ceramide C24:0, e sfingomielina
- Esteri del colesterolo C16:0 e C18:0
- Alterazioni simili nei topi SOD/SLA presintomatici e dopo stress ossidativo
- Altro sul CNS
- Tronco cerebrale: Perdita di neuroni motori: Risparmiati i nuclei extraoculari
- Corteccia precentrale: Perdita di cellule di Betz e altre piramidali
- Muscoli:
Inizialmente nel decorso
- Numerose piccole regioni di fibre muscolari atrofiche angolari
raggruppate
- Le fibre muscolari atrofiche in un gruppo sono spesso di vari
tipi.
- Pochi tipi raggruppanti eccetto che nei casi cronici, lentamente progressivi
- Trattamento
- Sintomatico
- Insufficienza respiratoria 16
- Ventilazione nasale-facciale
- Più efficace con funzione bulbare preservata
- Inizialmente notturna
- Criteri iniziali
- Correlata ai sintomi
- Ossimetria nel sonno: Saturazione dell'O2 < 88%, Per
cinque minuti continui
- Punto finale: Ossimetria senza integrazione di ossigeno
al 95% o maggiore
- Benefici: Può migliorare la sopravvivenza e la qualità della vita
- Prognosi: La maggioranza delle persone con la SLA che
iniziano una NPPV
non decidono di ricorrere alla tracheostomia
- Tracheostomia: Tipi elettivi
- Tracheostomia dilatazionale percutanea
- Apertura chirurgica
- Sicurezza
- Allenamento e esercitazioni per tutti i prestatori di cura
- Personale e corredo di rianimazione per il recupero
- Un secondo sistema ventilatorio: Per pazienti che
necessitano di ventilazione per 20
÷ 24 ore
- Evitare
- Somministrazione di ossigeno
- La pressione positiva continua alle vie aeree: Aumenta
il carico ai deboli muscoli inspiratori
- Tosse: Assistenza meccanica
- Riduzione dell'ansietà da dispnea: Lorazepam, sublinguale 0,5
÷ 1
mg
- Trattamento della dispnea terminale
- Morfina 2,5 ÷ 5 mg q4h
- Ansietà associata: Lorazepam 1
÷ 2,5 mg; Midazolam 1
÷ 2 mg
- Disfunzione bulbare: Gastrostomia
- Prima della gastrostomia: Cambiare la dieta con una
masticabile più facilmente, cibi altamente calorici
- La decisione per gastrostomia va basata sulla valutazione clinica, non
su quella radiologica
- Più efficace se condotta prima della compromissione respiratoria
- Benefici della gastrostomia
- Aumenta la sopravvivenza
- Migliora la qualità della vita
- Rischi della gastrostomia
- Aspirazione per eccesso di alimentazione
- Può richiedere intubazione o NPPV in pazienti con capacità vitale < 50%
- Crampi: Possono risolversi spontaneamente
- Solfato basico di chinina: 200 mg bid
- Carbamazepina: 200 mg bid
- Spasticità
- Baclofene in alcuni pazienti: 10
÷ 80 mg; Può aumentare
la debolezza
- Altro: Tizanidina; Memantina; Tetrazepam
- Raramente dantrolene: Solitamente aumenta la debolezza
- Affezione pseudobulbare
- Amitriptilina: 10 ÷ 150 mg
- Neurodex (Anavir): Destrometorfano (30mg) e Chinidina
(30mg) insieme
- Altri: Fluvossamina;
Litio; L-DOPA
- Debolezza: Apparecchi ortopedici, Supporti....
- Generale: Informazione e discussione
- Fornire una chiara diagnosi e informazione che la malattia è
progressiva nella maggior parte dei casi
- Permettere al paziente di mantenere una qualche speranza
- Incoraggiare lo sviluppo di sistemi di supporto
- Successivamente: Prendere contatto con l'ospizio
- Sopravvivenza
- Riluzolo
- Può aumentare la sopravvivenza con una media di 3 mesi
- ? Più efficace 15:
Insorgenza della malattia bulbare; Età più avanzata; Prima
nel decorso della malattia
- Non ha effetto su forza, respirazione o qualità della vita
- Costo alto: ~ $10.000 per anno
- Gastrostomia
- Supporto respiratorio
- Supporto dei prestatori di cura
- Collegamenti esterni: Linee guida pratiche della AAN
Disturbi simili alla SLA con demenza fronto-temporale
11
- Età dell'insorgenza: 4a ÷ 8a decade
- Clinica
- Demenza frontotemporale (FTD)
- Demenza
- Disturbi del linguaggio
- Cambiamenti di personalità
- Disturbi del comportamento
- Sindrome della sclerosi laterale amiotrofica (SLA)
- Disfunzione bulbare: Disfagia
- Denervazione degli arti
- Segni nei neuroni motori superiori negli arti
- Iperriflessia
- Spasticità meno preminente in alcuni pazienti
- Fascicolazioni: Possono avvenire senza segni di SLA
- Decorso e associazioni
- La SLA o la FTD possono presentarsi per prime
- Il tempo fra l'insorgenza delle sindromi può essere anni
- Più frequentemente la demenza si presenta per prima
- Associazione
- Il 14% dei pazienti con la FTD incontra i criteri per una SLA definita
- Il 36% dei pazienti con la FTD incontra i criteri per una SLA possibile
- Laboratorio
- EMG: Denervazione; Fascicolazioni
- Patologia del CNS
- Perdita neuronale nei lobi frontotemporali
- Inclusioni intraneuronali di ubiquitina immunoreattiva
- Frequenza
- Demenza SLA: 100%
- Pazienti SLA non diventati dementi: 20%
÷ 50%
- Demenza frontotemporale mancante dei sintomi motori: Alcuni pazienti
- Localizzazioni
- Cellule granulari dentate dell'ippocampo
- Neuroni nella II lamina della corteccia frontotemporale
(extramotoria)
- neuriti corticali distrofici neuriti corticali
- Neuroni motori
- Istochimica
- Ubiquitina: Colorazione
- Sinucleine tau e α: Nessuna Colorazione
- Corteccia: Astrocitosi (V lamina); Perdita di cellule di Betz
- Perdita di assoni del tratto piramidale: Più distalmente
- Nessuna evidenza patologica di altre condizioni dementizzanti
Disturbi del Pacifico occidentale simili alla SLA
6

- Eziologia
- ? Correlata a tossine ambientali nelle nocciole delle Cycadi (Cycas circinalis):
Possibilità di inclusione
di
- Tossina β-Metilamino-L-alanina (BMAA)
- Consumo di nocciole delle Cycadi contenenti la tossina BMAA
- Consumo di volpe volante Guamaniana: Concentra la BMAA ad
alti livelli 19
- Metilazossimetanolo (MAM): Agente alchilante, può produrre una malattia
con lunga latenza
- Altre associazioni ambientali
- Mancanza di calcio e magnesio
- Tau (MAPT)
:
Può essere un gene modificante aumentante il rischio per SLA-PD
- Mutazione TRPM
7
: Thr1482Ile
- Presente in alcuni pazienti SLA-Guam e PD-Guam
- Sclerosi Laterale Amiotrofica Parkinsonismo/Demenza complesso
1 (SLA-PD1)
- Epidemiologia
- Avviene in foci ad alta incidenza
- Isole Marianne e Guam del sud; penisola Kii del Giappone
- Frequenza in Guam nel 1960: Maschi 179/105; Femmine
60/105
- Raggruppamenti famigliari
- Chammoro
- Modelli di ereditarietà Mendeliana non chiari
- ? Dominante con penetranza ridotta
- Media dell'insorgenza: 52 anni
- Maschi:Femmine = 2:1 ÷ 3:1
- Cambiamenti recenti 13
- Diminuzione dell'incidenza: Riduzione da 8
÷ 10 volte
- Età avanzante dell'insorgenza
- Minore predominanza maschile
- Caratteristiche cliniche
- Malattia dei neuroni motori
- Caratteristica dei neuroni motori superiori ed inferiori: 90%
- Sindrome solamente dei neuroni motori inferiori: 10%
- Comune la caratteristica bulbare
- Altro sul CNS
- Parkinsonismo: 15%
- Demenza: 10%
- ? Segregate come malattia separata: Incorrono in altri membri della famiglia
- Retinite pigmentosa: 50%
- Durata della malattia
- Media: 4 anni
- Età alla morte: 48 ÷ 68 anni
- Storia famigliare positiva (SLA o PDC): 45%
- Patologia
- Groviglio neurofibrillario
- Localizzazione: Corteccia temporale mesiale; Gangli basali; Tronco cerebrale
- Contiene la proteina tau
- Corpi cellulari granulovacuolari
- Cellule del corno anteriore
- Perdita (100%)
- Inclusioni positive alla ubiquitina (50%)
Malattia atipica del neurone motorio con oftalmoplegia e
disturbi extrapiramidali 4
- Insorgenza
- 4a ÷ 7a decade
- Spesso disfunzione bulbare: Disartria
- Motorio
- Debolezza: Bulbare, braccia e gambe
- Segni nei neuroni motori superiori
- Presenti variabilmente
- Riflessi tendinei: Vivaci o normali
- Oculare
- Paralisi dello sguardo: Specialmente verticale; Sopranucleare
- Saccadi: Anormali
- Segni extrapiramidali: Variabili
- Rigidità
- Ballismo
- Tremori
- Progressione: Spesso rapida; Morte in 1 ÷ 3,5 anni
- Patologia
- Perdita delle cellule del corno anteriore: Con corpi cellulari di Bunina
- Alterazioni del tronco cerebrale: Dentato; Nuclei collicolari; Gliosi
grigia periacqueduttale
- Sostanza nera: Perdita neuronale
- Putamen e globo pallido: Perdita neuronale
Vedi anche
SLA ereditaria
Sclerosi Laterale Amiotrofica (SLA): Discussione della diagnosi
1
Obiettivi della consulenza iniziale
- Fornire informazione
- Informazioni appropriate su quello che il paziente vuole e può capire
- Informazioni appropriate sul grado di certezza della diagnosi di SLA
- Evitare l'imposizione di informazioni che il paziente non vuole
- Rispondere adeguatamente alla reazione del paziente all'informazione
Passi nella discussione
- Conduzione
- Area privata
- Evitare le interruzioni
- Presenza anche di parenti o amici del paziente
- Il medico è seduto a livello del paziente
- Stile della comunicazione: Diretta e empatica
- Scoprire out ciò che il paziente sa: "Cosa Le è stato detto
riguardo al problema?"
- Fornire le informazioni gradualmente: Piccoli pezzi di informazione con
gravità crescente
- Commento generale iniziale negativo: "Mi dispiace che i
risultati degli esami non siano come avevo sperato."
- Gerarchia descrittiva: Non dare messaggi molto pessimistici tutti in
una volta
- "La Sua debolezza è causata da un danno nei fili elettrici
(i nervi) che dicono ai muscoli di contrarsi"
- "I nervi si stanno gradualmente danneggiando"
- "La condizione è chiamata sclerosi laterale amiotrofica (SLA),
malattia di Lou Gehrig malattia o malattia dei neuroni motori"
- "Sfortunatamente, non ci sono cure, al momento."
- "Un farmaco, il Riluzolo può rallentare leggermente il problema."
- "Si, la malattia è spesso infine fatale, ma la sua progressione
in alcuni pazienti può essere lenta"
- La maggior parte dei pazienti sono più confortati da una "etichetta" per
la loro
condizione
- Rispondere adeguatamente alla reazione del paziente all'informazione
- Chiedere ai pazienti
- Se hanno avuto sufficiente informazione, o ne vorrebbero di più
- Se hanno altre domande
- Altre informazioni
- Brochure con informazioni sulla SLA in linguaggio non specialistico
- Organizzazioni che possono dare un ulteriore aiuto
- Accordo per ulteriori cure
- Appuntamento di controllo a breve termine
- Trattamento: Dipendente dallo stadio della malattia
- "Un farmaco, il Riluzolo può rallentare leggermente il problema."
- Terapia fisica e occupazionale
- Nutrizione
- Respiratorie
- Servizio di assistenza sociale: Per paziente e prestatori di
cura
- Risorse cliniche per aiutare il paziente nella lotta con la SLA
- La situazione degli studi e delle ricerche cliniche sulla SLA
- Enfatizzare la disponibilità per domande future
Protocollo creatina: Per SLA e disturbi neuromuscolari
- NOTE
- Ci sono alcune prove che questo trattamento produce un piccolo
miglioramento a breve termine nella forza (3%)
nelle
distrofinopatie e
nella
FSH.3
- C'è la prova che la creatina NON è di beneficio nella SLA
- Prima di iniziare un protocollo farmacologico: Misurare la forza con la
dinamometria quantitativa
- Drug Protocolli farmacologici
- Creatina monoidrato: Dissolta in liquidi con piccoli pasti
3
- Adulti: 10 grammi/giorno
- Bambini: 5 grammi/giorno
- Creatina monoidrato
- Giorni 1 ÷ 5: 12 grammi 3 volte al giorno
- Giorni 6 ÷ 30: 2 grammi una volta a giorno
- Creatina e carboidrato: Fosfageno HP
- Concentrazione: 5 grammi di creatina in 43 grammi di fosfageno (1
cucchiaio)
- Giorni 1 ÷ 5: 7 cucchiai per giorno
- Giorni 6 ÷ 30: 1 cucchiaio per giorno
- Dopo 30 giorni: Misurare la forza con la dinamometria quantitativa
Malattia del neurone motorio: Alterazioni alla MRI successivamente nel decorso
Segnale iperintenso alla MRI (FLAIR)
- Segnale focale nel tratto corticospinale, peduncolo cerebrale e capsula interna
- Le alterazioni alla MRI si riscontrano in alcuni ma non in tutti i pazienti
con la SLA
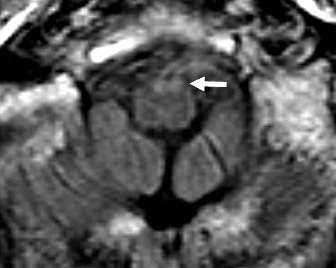
Midollo |
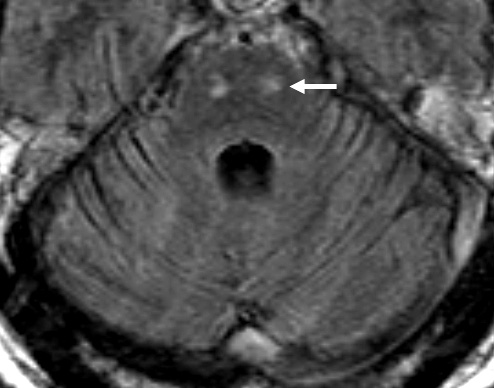
Ponte |
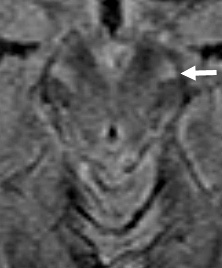
Cervello medio |
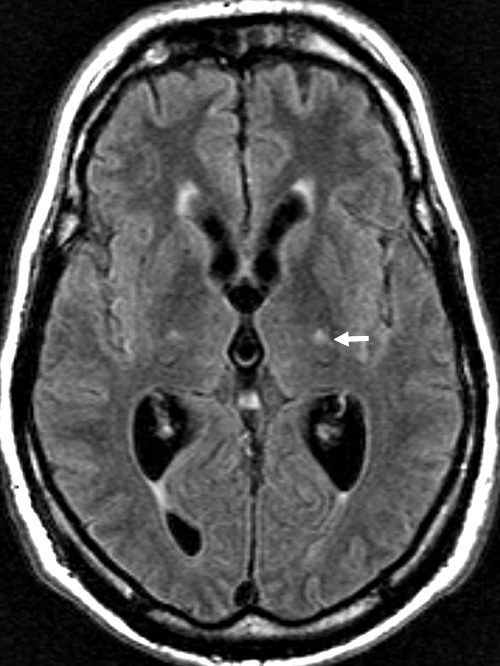
Capsula interna |
SLA: Storia iniziale del concetto
- 1830 Charles Bell: Riportò il caso di una donna di mezza età con
- Paralisi progressiva degli arti e della lingua
- Preservazione delle sensazioni
- Patologia: Porzione anteriore del midollo spinale mostrava degenerazione
- Conclusione: ""la colonna anteriore ...del midollo spinale è per
il movimento,
la colonna posteriore del midollo spinale è per le sensazioni...""
- 1850 Aran: Descrisse l'atrofia muscolare progressiva (PMA)
- Pensando che si trattasse di una malattia primaria dei muscoli
- 1855 Cruveilhier: Attribuì la PMA ad atopia del corno anteriore del midollo
spinale
- 1870 Fritsch e Hitzig: Identificarono la striscia reattiva motoria
corticale del cervello con la stimolazione elettrica
- 1874 Charcot: Stabilirono l'entità clinica della
Sclerosi Laterale Amiotrofica (SLA)
- Basandosi su 20 casi e 5 autopsie
- 1879 Kahler e Pick: Identificarono l'atrofia della corteccia motoria
nella SLA
- 1883 Dejerinne: Correlò la Paralisi Bulbare Progressiva (PBP) alla SLA
- 1884 Kahler: Raggruppò PMA, SLA e PBP come "degenerazioni primarie
del sistema
motorio"
 Informazioni
per i pazienti
Informazioni
per i pazienti
Muscular Dystrophy Associazione
3300 East Sunrise Drive
Tucson, AZ 85718
Telephone 520-529-2000
FAX 520-529-5300
SLA Associazione (National Office)
21021 Ventura Boulevard, Suite 321
Woodland Hills, CA 91364-2206 USA
Telephone 800-782-4747
SLA Society of Canada
220 - 6 Adelaide Street East
Toronto, Ontario, M5C 1H6 CANADA
Telephone 416-362-0269
Telephone 800-267-4257 (toll-free in Canada)
FAX 416-362-0414
e-mail: alssoc@inforamp.net
Altre informazione
Ritorna a Sindromi
motorie
Va alla SLA ereditaria
Ritorna a Neuromuscular home
page
Riferimenti
1. J Neurol Sci 1998;160:S127-S133
2. Disturbi neuromusculari 1999;9:593-597
3. Neurology 1999;52:854, Neurology 2000;54:1848-1850
4. Acta Neuropath 2000;100:342-346 1999;98:512-515
5. Neurology 2001;56:753-757
6. Brain 2001;124:2215-2222
7. J Neurol Sci 2001;191:3-9, Neurology 2002;59:280–282
8. Am J Hum Genet 2002;May
9. Neurology 2002;59:99-103
10. Neurology 2002;59:773-775, Am J Hum Genet 2004;75 On-line October
11. Neurology 2002;59:1077–1079, Acta Neuropathol 2002; July On-Line, Acta
Neuropathol 2002; October On-Line
12. Ann Neurol 2002;52:448-457
13. Am J Epidemiol 2003;157:149-157
14. Neurology 2003;60:1348–1350
15. J Neurol 2003;250:473–479
16. J NeurolSci 2003;209:1-4
17. J Neurol Neurosurg Psychiatry 2003;74:995–997
18. Nature Genet 2003; Online July
19. Neurology 2003;61:387–389, Neurology 2003;61:291–292
20. Neurology 2004;62:1753–1757
21. Am J Epidemiol 2004;160:26-33,
Neurology
2009;73:1693–1698
22. J Neurol Neurosurg Psychiatry 2006;77;388-389
23. Neurology 2006;67:1833–1836
24.
Ann Neurol 2007;61:427-434
25. Neurology 2008; Online January
26. Science 2008;319;20
27. J Neurol Sci 2008 Jun
11
28. Nat Clin Pract Neurol
2008;4:366-374
29. Amyotroph Laterale
Scler 2008 Jun 12:1-6
30. Amyotroph Laterale
Scler 2008 May 2:1-9
31. Neurology 2008; Online July
32. Am J Hum Genetica 2008; Online December
33. Hum Molec Genet 2009;
Online July 23
34. Nature Genet 2009;
Online September
35. Brain 2010 Oct 19
36. J Neurol Neurosurg
Psychiatry 2010 Nov 3
21 ottobre /2010
Va a Acronimi e sigle
Va a Mitolario in
italiano
Va a Mitolario italiano -
inglese
Va a Mitolario inglese -
italiano


{kind=link}