![]() Traduzione di Natale
Marzari
Traduzione di Natale
Marzari
Home,
Ricerca,
Indice alfabetico,
Collegamenti,
Patologia,
Molecole,
Sindromi,
Muscoli,
Gionezioni neuromuscolari,
Nervi,
Spinale,
Atassia,
Anticorpi e Biopsia,
Informazioni per i
pazienti

| Fondazione FONAMA | fonama@fonama.org | Telefono 335250742 |
|
|
|
Traduzione del 31 gennaio 2011
![]() Recenti prove dimostrano che numerose
sindromi di neuropatia motoria pura possono essere distinte
dalla sclerosi laterale amiotrofica (SLA). L'identificazione di queste
sindromi di neuropatia motoria è importante poiché, in contrasto alla SLA, esse sono
spesso mediate immunitariamente e trattabili.
Le sindromi di neuropatia motoria solitamente hanno caratteristici, ma non
oneici, modelli di debolezza e nessun segno nei motoneuroni superiori.
Solitamente sono necessari ulteriori esami di Laboratorio, compresi gli esami elettrodiagnostici e la misurazione
degli
autoanticorpi nel siero, per distinguere chiaramente questi disturbi
da altre
neuropatie demielinizzanti e sindromi dei motoneuroni.
Recenti prove dimostrano che numerose
sindromi di neuropatia motoria pura possono essere distinte
dalla sclerosi laterale amiotrofica (SLA). L'identificazione di queste
sindromi di neuropatia motoria è importante poiché, in contrasto alla SLA, esse sono
spesso mediate immunitariamente e trattabili.
Le sindromi di neuropatia motoria solitamente hanno caratteristici, ma non
oneici, modelli di debolezza e nessun segno nei motoneuroni superiori.
Solitamente sono necessari ulteriori esami di Laboratorio, compresi gli esami elettrodiagnostici e la misurazione
degli
autoanticorpi nel siero, per distinguere chiaramente questi disturbi
da altre
neuropatie demielinizzanti e sindromi dei motoneuroni.
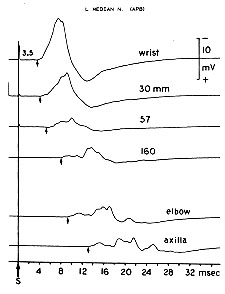
Un rapporto patologico di Rowland e di altri documentò per primo che il sito primario della malattia di un paziente con una sindrome motoria pura potava essere lungo il corso degli assoni. Questo paziente, con una sindrome dei motoneuroni inferiori (LMN) e una proteina M nelle IgM del siero, aveva un danno agli assoni motori ma non ai corpi cellulari. Le neuropatie motorie venivano inizialmente diagnosticate durante la vita con una esaminazione elettrodiagnostica. Gli studi di conduzione nervosa mostrarono il blocco degli impulsi in siti focali lungo il corso degli assoni motori (blocco di conduzione motoria) fornendo una forte prova che il sito primario della malattia si trova nei nervi periferici piuttosto che nelle cellule del corpo. Il fenomeno del blocco di conduzione era stato descritto precedentemente in pazienti con neuropatie moto-sensorie (polineuropatia infiammatoria Demielinante cronica (CIDP)). Si pensò che il blocco di conduzione originava dalle regioni focali di demielinazione mediata immunitariamente lungo il percorso del nervo.
Nel 1986 venne riportato di un paziente con una sindrome LMN senza blocco di conduzione, ma con una proteina M nel siero IgM che legava alle GM1 gangliosidiche. In questa circostanza l'associazione della sindrome motoria con un autoanticorpo diretto contro un antigene neurale suggeriva che il disturbo poteva essere mediato immunitariamente. Comunque, i tentativi con una immunosoppressione non ebbero effetto sulla progressione della malattia in quel paziente. Una risposta clinica all'immunoterapia rimane a "uno standard d'oro", senza il quale è difficile arguire che una sindrome è mediata immunitariamente. Nel 1988 venne riportato che due pazienti con una neuropatia motoria multifocale, blocco di conduzione motoria, e anticorpi IgM anti-GM1 nel siero migliorarono dopo il trattamento con ciclofosfamide. Appare ora che sia il blocco di conduzione motoria sia gli anticorpi anti-GM1 nel siero da soli possono essere marcatori per pazienti con sindromi LMN che spesso migliorano dopo le terapie immoneomodulanti.
|
|
Blocco di conduzione Il quantitativo del blocco aumenta con la stimolazione più prossimale. |
2) I pazienti con sindromi dei motoneuroni inferiori distali (D-LMN) hanno, per definizione, perdita di assoni motori ma nessun blocco di conduzione. Comunque, essi sono clinicamente simili ai pazienti con la MMN, con debolezza asimmetrica lentamente progressiva che più comunemente inizia nelle braccia. In molti pazienti con sindromi D-LMN gli studi elettrofisiologici non forniscono un aiuto definitivo per la distinzione fra neuropatie motorie mediate immunitariamente dalle malattie dei motoneuroni intrattabili. Comunque, in alcune pazienti con sindromi D-LMN si ha la dispersione temporale della composizione dei potenziali di azione muscolari, anormalità delle onde F e altre alterazioni suggerenti la demielinazione. Anche prove di lieve demielinazione segmentale nella biopsia nervosa, sebbene non siano diagnostiche, possono fornire un indizio che una sindrome D-LMN è mediata immunitariamente.
Galβ1-3GalNAcβ1-4Galβ1-4Glcβ1-1'Ceramide
3
|
Neu5Acα2
|
Un problema maggiore che riguarda l'esaminazione clinica per anticorpi anti-GM1 sono le differenze nella metodologia usata nella loro misurazione. La tecnica della metodologia di validazione nella identificazione dei sieri con alto titolo anticorpale non è sufficiente. I laboratori devono anche documentare la sensibilità e la specificità degli anticorpi anti-GM1 con metodi di esaminazione con studi di correlazione clinica usando sieri da pazienti con sindromi motorie.
Nel nostro Laboratorio la sensibilità dell'esaminazione degli anticorpi (IgM vs Co-GM1 e NP-9)
per la MMN è ora 85% ÷ 90%. In generale, un alto titolo
dio anticorpi IgM anti-GM1 nel siero dovrà essere trovato in:
1. Almeno 80% dei pazienti con neuropatie motorie multifocali, e
2. Meno del 1% dei pazienti con sclerosi laterale amiotrofica tipica.
Una comune pratica ambigua è la citazione, in rapporti di esami, di statistiche dalla
letteratura senza la validazione clinica dei metodi specifici usati in un
Laboratorio. I laboratori che non possono fornire una correlazione dei dati,
della relazione della sindrome del paziente con i risultati della loro specifica metodologia, non sono qualificati a condurre misurazioni degli anticorpi serici anti-GM1 come esame clinico.
Il miglioramento nella forza dopo un trattamento con
HIG (per esempio 1 g/kg/giorno x 2 giorni) è comune (50%
÷ 70% dei casi),
ma la durata del beneficio è variabile, può durare 2 settimane fino a 6 o più mesi.
La dose e la frequenza dei trattamenti successivi è basata sulla risposta individuale del paziente.
1. Dopo un trattamento con HIG dovrà essere monitorato il periodo di massimo
miglioramento. Successivamente si dovranno fare i trattamenti appena prima di
quando ci si aspetta una ricaduta. La
dose minima efficace delle HIG può essere determinata dalla riduzione sequenziale
delle dosi
successive di HIG del 10% fino a quando si raggiunge un livello che produce un
beneficio minore in qualcosa (tempo di durata, o grado, di miglioramento della forza).
La dose minima che produce un miglioramento ottimale sarà quella da usare poi
nella terapia a lungo termine.
2. La mancanza del miglioramento nella forza dopo uno, o al più due, trattamenti (totale 2
÷ 3 g/kg
ciascuno) dovrà essere considerata un trattamento insufficiente. Non si dovrà
usare nessuna ulteriore HIG.
Sebbene gli effetti collaterali delle HIG siano solitamente benigni, il suo grande costo impone
l'obiettiva documentazione di ogni beneficio, compresi l'esaminazione quantitativa
dei muscoli e
la valutazione funzionale, per giustificare la continuazione del loro uso. Sono necessari
anche ulteriori studi per documentare se il trattamento con le HIG del sottostante processo patogeno
nella MMN produce
un beneficio sintomatico mentre consente al sottostante processo immunitario di progredire.
Il trattamento con corticosteroidi (Prednisone o Solumedrolo) è raramente utile
nella MMN e può spesso esacerbare la debolezza.
Il ciclofosfamide è il solo farmaco immunosoppressore che sia stato riportato indurre
un beneficio a lungo termine
in molti pazienti (50% ÷ 80%) con la MMN. Sfortunatamente, la sua tossicità,
specialmente l'aumento rischio di neoplasia con alte dosi cumulative in tutta la vita (>75 g), richiede una attenta analisi
del rapporto rischio:beneficio in ciascun
paziente. Il regime terapeutico utilizzerà dosi di ciclofosfamide che siano
sufficientemente alte per ridurre il titolo anticorpale anti-GM1 del 60%, o più. Noi originalmente
usavamo una dose iniziale di 3 g/M2 per 8 giorni seguita da una somministrazione orale
continuativa di
(100-150 - mg/giorno di per 6 ÷12 mesi).
L'esperienza più recente suggerisce che 6 trattamenti al mese con ciclofosfamide
endovenoso (1g/M2), ciascuno preceduto da due scambi del plasma, hanno la stessa efficacia, hanno
minori effetti collaterali e utilizzano una dose cumulativa del farmaco 50%
÷ 70% minore. Questo regime produce a sostenuta
riduzione nel titolo degli anticorpi serici anti-GM1 in approssimativamente 60%
÷ 80% dei pazienti. La maggior parte dei pazienti nei quali
il titolo anticorpale si riduce mostrano un beneficio funzionale. La remissione solitamente persiste per 1-3 anni; dopo
dei quali,
il titolo anticorpale spesso aumenta e la debolezza ricorre. In questo caso può
essere necessario ripetere il trattamento.
La decisione sull'opportunità di trattare i pazienti con sindromi D-LMN
senza la prova elettrodiagnostica della demielinazione può essere difficile.
Un titolo alto degli anticorpi serici IgM anti-GM1 è un utile indicatore che una sindrome D-LMN può essere mediate immunitariamente e trattabile.
Anche la prova di demielinazione alla biopsia del nervo surale può essere utile a questo riguardo.
Un miglioramento misurabile nella
in forza dopo il trattamento con HIG può fornire un supporto per una ulteriore
immunoterapia, con agenti come il ciclofosfamide o una ulteriore infusione periodica
di HIG.
Neuropatie motorie e disturbi dei motoneuroni. Alcune neuropatie motorie erano state classificate come varianti SLA, con predominanza di segni LMN e alterazioni assonali agli esami elettrodiagnostici. Certe caratteristiche possono aiutare nella differenziazione tra neuropatie motorie e SLA. I pazienti con neuropatie motorie possono avere preservazione dei riflessi nei muscoli deboli, ma la spasticità aperta e le caratteristiche bulbari sono cospicuamente mancanti. Questo è in contrasto con i pazienti con la SLA i quali spesso hanno risultanze preminente dei motoneuroni superiori e bulbari. Anche il decorso prolungato che viene spesso notato nei pazienti con neuropatie motorie aiuta a differenziare le loro sindromi dalla SLA tipica. Le neuropatie motorie acquisite producono più spesso debolezza asimmetrica. Questo modello è solitamente clinicamente distinto dalla debolezza prossimale simmetrica che caratterizza la maggior parte delle atrofie muscolari spinali ereditarie.
Diverse sindromi dei motoneuroni inferiori che erano state descritte come di eziologia incerta potrebbero essere disturbi degli assoni motori o delle cellule del corpo. Una maggioranza dei pazienti con sindromi D-LMN non hanno ne prove di demielinazione dei nervi periferici ne anticorpi anti-gangliosidi nel siero. Questi pazienti tendono ad avere debolezza più rapidamente progressiva di quella che è tipica per le neuropatie motorie mediate immunitariamente . Contrariamente alla SLA tipica, molti pazienti con la D-LMN non sviluppano mai la disfunzione bulbare. Non ci sono rapporti di risposte al trattamento immunosoppressivo in pazienti con la D-LMN ne con demielinazione ne con autoanticorpi nel siero. Alcuni pazienti sviluppano una sindrome motoria inferiore asimmetrica progressiva con debolezza predominante inizialmente nella muscolatura prossimale (sindromi P-LMN). Le caratteristiche cliniche comprendono esordio a tarda età, predominanza maschile (85%) e iniziali segni di debolezza nelle estremità superiori (80%). La progressione è lenta. La debolezza è spesso confinata ad una o due estremità per 3 ÷ 5 anni. Gli studi elettrodiagnostici mostrano solamente prove di perdita assonale. Alcuni pazienti con sindromi P-LMN (30%) hanno anticorpi serici selettiva leganti agli GA1 gangliosidici. Comunque, non ci sono prove che le sindromi P-LMN rispondano al trattamento immunosoppressivo .
Amiotrofia monomelica, una sindrome che colpisce principalmente giovani (15 ÷ 25 anni) maschi (80%), si presenta con debolezza della muscolatura distale ad una delle estremità superiori che progredisce per 1 ÷ 2 anni e poi rimane stabile. Occasionali pazienti sviluppano debolezza nell'arto opposto, lievi sintomi sensori o tremore. Gli studi elettrodiagnostici mostrano denervazione negli gli arti colpiti. Non c'è associazione con anticorpi serici.
Sono stati riportati rari
pazienti con la sindrome LMN
paraneoplastica. Il caso meglio descritto
fra questi è una
neuronopatia motoria subacuta
associata con linfomi, come nella malattia di Hodgkin.,, più gravemente nelle gambe, A volte quando la neoplasmia è in remissione o durante
l'irradiazione sviluppano una debolezza asimmetrica progressiva. Raramente la debolezza è grave, e
spesso si stabilizza o migliora in un periodo di mesi o anni. Gli studi patologici
mostrano una perdita di neuroni motori nel corno ventrale del midollo spinale e
qualche
coinvolgimento del tratto sensorio. Il coinvolgimento della LMN è stato anche ben descritto
costituire parte occasionale della encefalomielite paraneoplastica e delle
sindromi della gangliopatia sensoria che si hanno in associazione con gli anticorpi anti-Hu. Non ci
sono prove chiare che ci sia un aumento nell'incidenza delle sindromi paraneoplastiche
della SLA "tipica", con coinvolgimento dei motoneuroni ed inferiori. E' stato
riportato che alcune pazienti con la sindrome
simile alla SLA e neoplasmie, comprese delle cellule renali, del polmone e linfoma, erano migliorati o stabilizzati dopo
il trattamento del cancro.
Neuropatie demielinizzanti immunitarie. Sebbene
la MMN e
la
CIDP siano entrambe neuropatie
demielinizzanti, le differenze nelle loro caratteristiche cliniche,
elettrofisiologiche e immunologiche sono maggiormente preminenti delle loro
similarità. La MMN comunemente si presenta con debolezza distale asimmetrica
mentre nella CIDP, la risultanza più comune è la debolezza prossimale
simmetrica. Le remissioni e le ricadute nel decorso che si possono avere nella
CIDP sono insolite nelle neuropatie motorie. I pazienti con una MMN raramente
hanno sintomi sensori significativi mentre nella CIDP, i segni sensori sono la
regola. L'esaminazione elettrofisiologica può mostrare blocco di conduzione in entrambe condizioni.
Comunque, altre caratteristiche della demielinazione come la latenza distale prolungata e
il rallentamento della velocità di conduzione sono maggiormente preminenti nella CIDP. Anormalità
negli studi di conduzione nervosa sensoria si vedono solitamente nella CIDP, ma
non nella MMN, se non complicata da un altro processo di malattia.
L'esaminazione del liquido spinale mostra marcatamente aumentata la concentrazione della proteina nella maggioranza
dei casi di CIDP mentre questo cambiamento è raro nei pazienti con la MMN. Un titolo alto
degli anticorpi anti-GM1 come pure più specifici modelli di reattività agli autoanticorpi (vedi al di sopra) sono comuni in MMN.
Nella CIDP gli anticorpi anti-GM1 sono
insoliti. Autoanticorpi serici legante alla tubulina sono più comuni . Infine,
le differenze nella frequenza della risposta terapeutica al prednisone e lo scambio del plasma (comune
nella CIDP, ma raro nelle neuropatie motorie) definisce una
differenza pratica nella gestione dei due disturbi.
Altre neuropatie demielinizzanti mediate immunitariamente hanno un maggiore coinvolgimento sensorio e vengono raramente confuse con la MMN. Le neuropatie anti-MAG sono spesso associate con la debolezza, ma la perdita sensoria è la caratteristica di presentazione più frequente, e più disabilitante della malattia. Le neuropatie con anticorpi anti solfatide e la sindrome GALOP hanno persino un coinvolgimento sensorio più predominante. La sindrome POEMS può produrre grave debolezza ma questa è accompagnata da preminente perdita sensoria e da segni sistemici.
Durante i successivi 10 mesi ci fu debolezza progressiva e perdita di riflessi a dispetto della terapia con alte dosi di prednisone e 11 trattamenti con scambio del plasma. Vennero eseguiti sei trattamenti ogni mese con due scambi del plasma seguiti da ciclofosfamide endovenose (1 gm/M2 ) con miglioramento ad iniziante dopo 3÷4 mesi, e progredente a forza pressoché normale nell'anno successivo. Lei rimase stabile, senza alcun farmaco, per 3 anni. Lei poi notava lieve debolezza ricorrente nella mano destra in una distribuzione simile a quella all'esordio della malattia 5 anni prima.
COMMENTO: Una debolezza asimmetrica, sviluppante distalmente in un braccio o una mano, è il modello più comune di coinvolgimento iniziale nella MMN. I riflessi sono spesso normali all'inizio del decorso della malattia. I segni sensori significativi sono rari, ma i pazienti occasionalmente notano sintomi come parestesie, o persino anormali sensazioni del gusto. Il trattamento con il prednisone è raramente efficace, e è spesso associato con rapida esacerbazioni della debolezza. La decisione di usare il ciclofosfamide venne presa solamente quando 1. divenne chiaro che la paziente aveva sviluppato una significante disabilità, e 2. c'erano chiari segni che era presente un disturbo immunitario , comprendente un blocco di conduzione e un titolo serico alto degli anticorpi IgM anti-GM1. Il miglioramento della forza dopo il ciclofosfamide inizia successivamente, spesso 3 ÷ 6 mesi dopo l'inizio della terapia, e il decorso continua fino ad un anno dopo la fine del trattamento.
2° caso: Un uomo di 52 anni notava il piede destro ricadente. Nei 6 mesi successivi la debolezza ed i crampi peggiorarono progressivamente nella gamba destra e si svilupparono anche nella gamba sinistra. All'esaminazione risultò una debolezza asimmetrica, predominantemente nelle gambe. il tono muscolare era normale. I nervi cranici erano normali. I riflessi tendinei erano assenti alla caviglia destra, ma 2+ altrove. La sensazioni era normale. L'esame elettrodiagnostico mostrò denervazione nei muscoli paraspinosi del torace e ad entrambe le estremità inferiori. Le velocità di conduzione nervosa erano normali. Non vennero trovati anticorpi serici anti-GM1.
COMMENTO: Questo paziente ha una debolezza relativamente rapidamente progressiva con prove di coinvolgimento dei neuroni motori inferiori, ma non dei superiori. Il decorso clinico è più rapido di quanto si vede di solito nella MMN. Le categorie diagnostiche più appropriate saranno il termine più vecchio, “Atrofia muscolare progressiva”, o, descrittivamente, “sindrome dei motoneuroni inferiori”. Tali pazienti possono non sviluppare segni bulbari, o dei motoneuroni superiori, e mai soddisfare i criteri diagnostici per SLA. La denervazione nei muscoli paraspinosi del torace è più suggestiva di una malattia dei motoneuroni che di una neuropatia motoria. Senza prove di demielinazione o anticorpi serici che suggeriscano una eziologia immunitaria per la sindrome, è probabile che in questo paziente vedrà continuare la progressione di una debolezza che non risponde ai trattamenti immunosoppressivi..
3° caso: Ad un uomo di 55 anni era stata prescritta la possibilità di una immunosoppressione per trattare una sindrome dei motoneuroni inferiori con anticorpi anti-GM1. Lui aveva notato una aumentante difficoltà a salire le scale e ad alzarsi dalla sedia 10 ÷ 15 anni. Negli anni recenti il suo parlare era diventano smozzicato ed aveva bisogno di un tempo più lungo per mangiare. Alla esaminazione generale risultò una lieve ginecomastia. L'esaminazione neurologica mostrò debolezza e fascicolazioni della lingua e della faccia. La lingua mostrò una grave atrofia. Era presente una moderata debolezza prossimale simmetrica. I riflessi tendinei erano difficile da provocare. Le sensazioni erano ridotte i tutte le modalità distalmente nel piedi. L'esame elettrodiagnostico mostrò denervazione cronica, più preminente nella faccia, nella lingua, e nei muscoli prossimali. La ripetizione dell'esaminazione degli anticorpi anti-GM1 mostrò un modello di IgM seriche polireattive leganti agli GM1 gangliosidici e all'istone H3 ad un titolo di circa 1.500.
COMMENTO: La debolezza del paziente era prossimale e simmetrica, più tipica dei disturbi dei motoneuroni ereditari che delle neuropatie motorie acquisite. Sebbene il paziente avesse un alto titolo degli anticorpi anti-GM1, il modello del legante era polireattivo. Gli anticorpi polireattivi non sono specifici per le sindromi motorie immunitarie, e vengono trovati anche nel 3% ÷ 5% pazienti con la SLA e le atrofie muscolari spinali ad esordio nell'età adulta. Una ulteriore esaminazione rivelò un eccessivo numero di ripetizioni trinucleotidiche nel recettore per gli androgeni, una risultanza coerente con una atrofia muscolare bulbo-spinale ereditaria collegata a X. Questo caso enfatizza che la determinazione della specificità degli anticorpi anti-GM1 è di aiuto per determinare la loro rilevanza clinica. Gli anticorpi polireattivi in un paziente con una neuropatia motoria con caratteristiche atipiche non erano non, di per se stesse, una indicazione per una terapia immunosoppressiva..
Lopate G, Pestronk A. Cronica Immunitaria Demielinante neuropatie. Seminars in
Neurology 1994;14:131-136.
Diagnosi differenziale di Demielinante neuropatie, incluso MMN
Parry G. Neuropatia motoria con Multifocali blocco di conduzione. In: Dyck PJ,
Thomas PK, Griffin JW, Bassa PA, Poduslo JF, eds. Neuropatia periferica. 3a ed.
Philadelphia: W.B. Saoneders Company 1993 pp 1518-1524.
MMN con un emphasis on the electrodiagnostic caratteristiche
Pestronk, A., Choksi, R. Neuropatia multifocale motoria: Siero IgM anti-GM1
ganglioside anticorpi nella maggior parte pazienti detected usanti covalente linkage di GM1
a ELISA plates. Neurology 1997;49:1289-1292.
Pestronk, A, Choksi R, Blume, G, Lopate, G. Neuropatia multifocale motoria: Siero
IgM associazione a a GM1-ganglioside contiene lipidi Miscela ma non a GM1 alone.
Neurology 1997;48:1104-1106.
Pestronk A, Lopate G, Kornberg AJ, et al. Distale lower motoneurone sindrome
con alti-titer siero IgM anti-GM1 anticorpi -miglioramento a seguito
immoneotherapy con mensili plasma scambio e intravenoso cyclophosphamide.
Neurology 1994;44:2027-2031.
Trattamento per lower motoneurone sindromi e MMN usanti lower cumulative Dosi
di intravenoso cyclophosphamide.
Pestronk A, Chaudhry V, Feldman EL, et al. inferiori motoneurone sindromi
definita da modelli di Debolezza, nervo conduzione Anomalie, e alti titers
di antiglycolipid anticorpi. Ann Neurol 1990;27:316-326.
Detailed descrizione di differenti lower motoneurone sindromi e associate
autoanticorpi.
Takigawa T, Yasuda H, Kikkawa R, Shigeta Y, Saida T, Kitasato H. anticorpi
contro GM1 ganglioside affect K+ e Na+ currents in isolated rat mielinizzati
fibre nervose. Ann Neurol 1995;37:436-442.
Pathogenic effetti di anti-GM1 anticorpi.
|
Ritorno a Fonama.org Home Page |
Alla pagina originale
|
|