|
La biologia mitocondriale è una delle aree a più rapida crescita in genetica
e in medicina, connettendo discipline scientifiche andanti dalla embriologia al cancro, alla malattia infettiva.
Sicuramente, si sa ora che i disturbi
del metabolismo mitocondriale giocano un ruolo non solo in rare malattie della fanciullezza , ma sono implicate anche in molte comuni malattie dell'invecchiamento,
includendo la malattia cardiaca, il diabete, la malattia di Parkinson e la demenza.
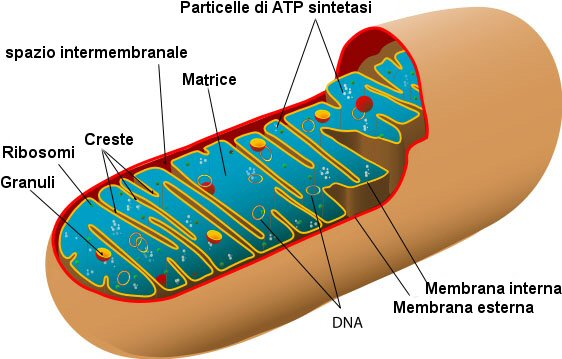
Centrali energetiche e molto di più
Il detto convenzionale in biologia ed in medicina
è che mitocondri funzionino solo come "centrali energetiche" per le cellule. Questo
eccesso di semplificazione è un errore che ha rallentato
il nostro
progresso verso la comprensione della biologia sottostante la malattia mitocondriale.
Occorrono circa 3000 geni per fare un mitocondrio.
Il DNA mitocondriale codifica solamente 37 di questi geni; i rimanenti geni sono
codificati nel nucleo delle cellula e le proteine risultanti sono trasportate nei mitocondri. Solamente
circa il 3% del geni sono necessari ed utilizzati dal
mitocondrio (100 dei 3000) per fare l'ATP. Più del 95% (2900 di 3000) sono coinvolti
in altre funzioni legati ai
compiti specializzati delle cellule differenziate nelle quali essi risiedono.
Questi compiti cambiano durante lo sviluppo dall'embrione all'adulto, e
man mano che i nostri tessuti
crescono, maturano, e si adattano allo sviluppo post-natale. Queste altre
funzioni non correlate all'ATP sono intimamente coinvolte con la maggior parte dei maggiori percorsi
metabolici usati dalle cellule per formare, demolire, e riciclare i
blocchi
molecolari che le costituiscono. Le cellule non sono nemmeno in
grado di fare gli RNA e DNA che necessitano loro per crescere e
funzionare senza i mitocondri. I blocchi costitutivi degli RNA e DNA sono
le purine e le pirimidine. I mitocondri
contengono gli enzimi limitanti la velocità per la biosintesi delle pirimidine
(diidroorotato deidrogenasi) e la sintesi finale (acido d-amino levulinico
sintetasi) necessaria per fare l'emoglobina. Nel fegato, i mitocondri
sono specializzati per detossificare l'ammoniaca nel ciclo dell'urea. I mitocondri
sono anche necessari per il metabolismo del colesterolo, per la sintesi degli estrogeni e
del testosterone, per il metabolismo dei neurotrasmettitori, e per la
produzione e detossificazione dei radicali liberi. Essi fanno
tutto questo in aggiunta alla demolizione (ossidizzazione) dei grassi,
delle proteine, e dei carboidrati che noi mangiamo
e beviamo.
Definizione di malattia mitocondriale
Le malattie mitocondriali sono il risultato di
mutazioni sia ereditate o spontanee nel mtDNA o nDNA le quali
portano ad alterate funzioni delle proteine o delle molecole di RNA che normalmente
risiedono nei mitocondri. I problemi della funzione mitocondriale,
comunque, possono colpire anche solo certi tessuti come risultato di fattori
subentranti
durante lo sviluppo e la crescita che noi non comprendiamo ancora. Persino
quando si prendono in considerazione le isoforme tessuto-specifiche
delle proteine mitocondriali, è difficile spiegare la variabilità dei
modelli dei sistemi d'organo colpiti nelle sindromi delle malattie mitocondriali viste clinicamente.
Genocopie della malattia mitocondriale
Poiché
i mitocondri
conducono così
tante differenti funzioni in tessuti diversi, ci sono letteralmente
centinaia di differenti malattie mitocondriali. Ogni disturbo produce
uno spettro di anormalità che possono essere confondenti sia per i pazienti
che per i
medici nei primi stadi di diagnosi. Poiché le complesse interazioni fra
le centinaia di geni e cellule che devono cooperate per mantenere nostro
oliati e funzionanti i nostri meccanismi metabolici, è un marchio caratteristico delle malattie mitocondriali che identiche mutazioni del mtDNA possono non produrre
identiche malattie. Le genocopie sono malattie che sono causati
dalle stesse mutazioni ma che possono non avere lo stesso aspetto clinicamente.
Fenocopie della malattia mitocondriale
E' vero anche il rovescio: differenti mutazioni negli mtDNA e nDNA possono portare alla stessa malattia. In genetica, queste sono conosciute come fenocopie. Un buon esempio è
la sindrome di Leigh, la quale può essere causata
da circa una dozzina differenti difetti dei geni. La sindrome di Leigh, originalmente
una descrizione neuropatologica del cervello di un bambino colpito, che
venne
descritta da Denis Leigh, il distinto medico britannico nel
1951. E' caratterizzata da anormalità bilateralmente simmetriche
alla MRI nel tronco cerebrale, cervelletto, e gangli basali, e spesso
accompagnate da elevati livelli dell'acido lattico nel sangue o nel liquido cerebrospinale.
La sindrome di Leigh può essere causata dalle mutazioni NARP, dalla mutazione MERRF,
da carenza del complesso I, dalla carenza di citocromo ossidasi (COX),
dalla carenza di piruvato deidrogenasi (PDH), e altre alterazioni non
mappate del DNA. Comunque, non tutti i bambini con queste anormalità del DNA svilupperanno
la sindrome di Leigh.
Le malattie mitocondriali sono persino più complesse negli adulti poiché
con l'invecchiamento si possono riscontrare alterazioni del mtDNA e,
all'incontrario, il processo di invecchiamento in se stesso può causare
dei deterioramenti della unzione mitocondriale.
C'è un largo spettro di disturbi metabolici, ereditati e acquisiti
in adulti nei quali è stata postulata o dimostrata un anormale funzione
mitocondriale.
adattato/selezionato da una sezione Robert Naviaux's "Panoramica, lo spettro
della malattia mitocondriale" nella
Mitocondriali e
Metabolic Disturbi, primario Care Guida per medici, prima
edizione. |













