| Fondazione FONAMA | fonama@fonama.org | Tel. 335250742 |
 |
 |
Traduzioni a cura di Natale Marzari Dopo 41 anni e 5 mesi, nel maggio 2006 la magistratura di Trento ha riconosciuto l'esistenza e la gravità di quella malattia rara che nessuna altra istituzione o persona singola della provincia di Trento ancora mi riconosce, e per negare la quale ancora mi perseguita. Natale Marzari |
||
MALATTIE MITOCONDRIALI e METABOLICHE UNA GUIDA PER MEDICI DI FAMIGLIA INTRODUZIONE nel trattamento dei bambini
con malattia mitocondriale Le malattie mitocondriali possono essere confuse nella diagnosi e nella loro gestione complessiva. Il medico di famiglia agisce come cuore essenziale del team di trattamento multidisciplinare. Quando viene sospettata una malattia mitocondriale, il medico di famiglia prescrive le iniziali procedure diagnostiche e implementa un trattamento ad interim intanto che viene deciso a chi rivolgersi e dove. Anche se il bambino viene mandato da uno specialista o in un centro, il medico di famiglia deve comunque rimanere il gestore del suo caso medico. I genitori devono aspettarsi regolari, via via comunicazioni dallo specialista, e una volta ottenute, coordinare e condividere le informazioni con la scuola, i terapisti, e gli altri agenti coinvolti. Inoltre, il medico di famiglia dovrebbe essere in grado di distinguere le normali malattie infantili e le crisi che meritano ulteriori consultazioni con lo specialista, mentre monitora la crescita e lo sviluppo, lo stato nutrizionale, ed il generale benessere del bambino. Lo specialista può anche chiedere al medico di famiglia di monitorare specifici parametri biochimici e effetti collaterali dei trattamenti.
Il medico di famiglia forma uno stretto legame con la famiglia ed il bambino,
"in-formandoli" in modo che essi possano diventare componenti attive del team.
Il medico di famiglia deve prendersi il tempo di ascoltare e rispondere
alle preoccupazioni della famiglia , fornendo materiale informativo e
indirizzandoli ai gruppi di supporto ed ai servizi sociali. Il medico di
famiglia deve inoltre assicurarsi che la famiglia riceva accurate e
puntuali/regolari informazioni che essi possano sul trattamento del
bambino, sui progressi, e nuove scoperte scientifiche che possano
influire sul protocollo del trattamento del bambino. Il patrocinio
con le compagnie di assicurazione è un altro essenziale componente nella cura di una
bambino con una La medicina mitocondriale è un campo di studio nuovo e in rapido sviluppo. Questo particolare opuscolo viene distribuito ai pediatri di base con l'intenzione di fornire alcune informazioni basilari sulle malattie mitocondriali ed un utile elenco di risorse dove poter attingere ulteriori informazioni. |
UNA GUIDA PER MEDICI DI FAMIGLIA
PANORAMICA
La gamma delle
malattie mitocondriali Robert K. Naviaux, MD,PdH
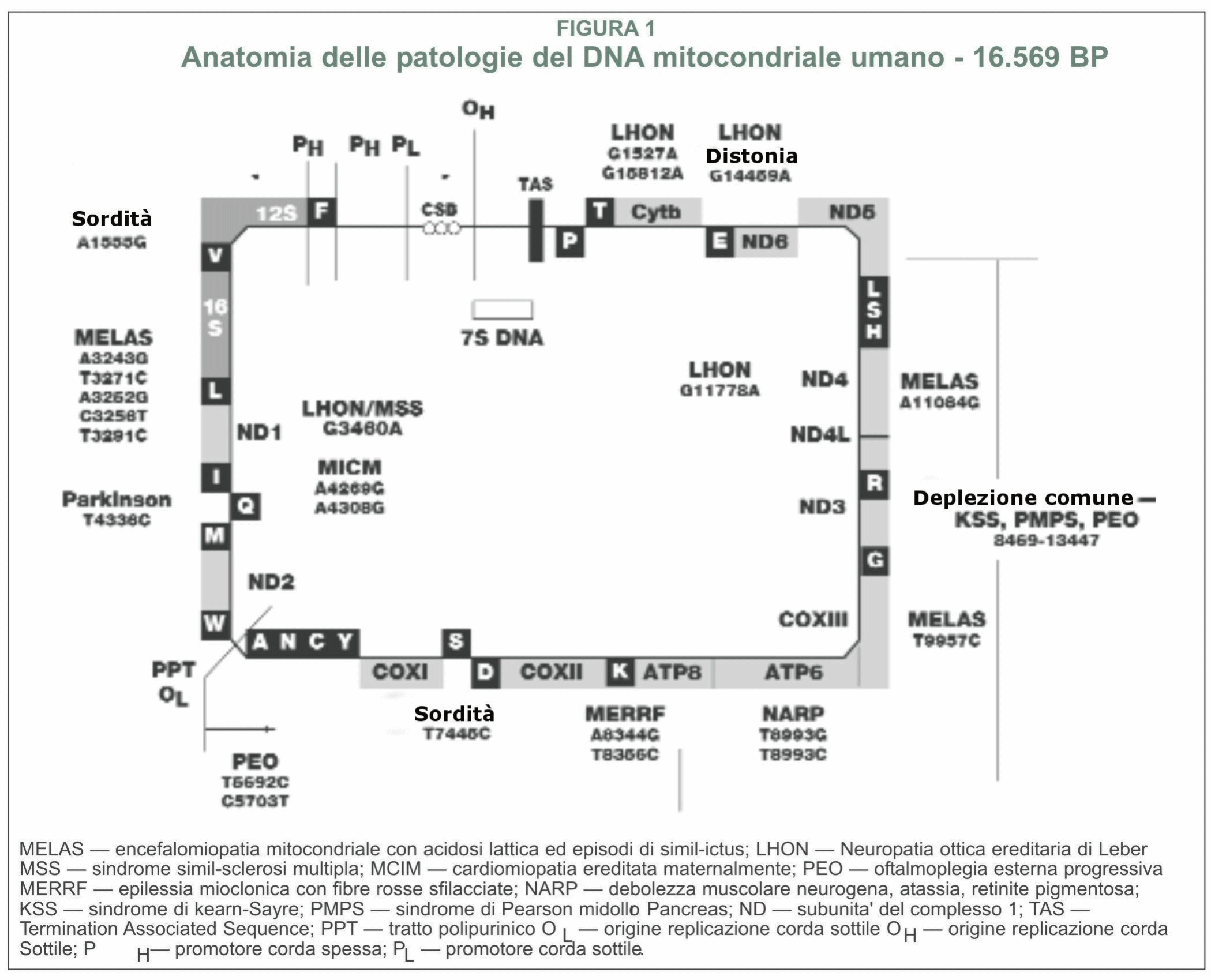
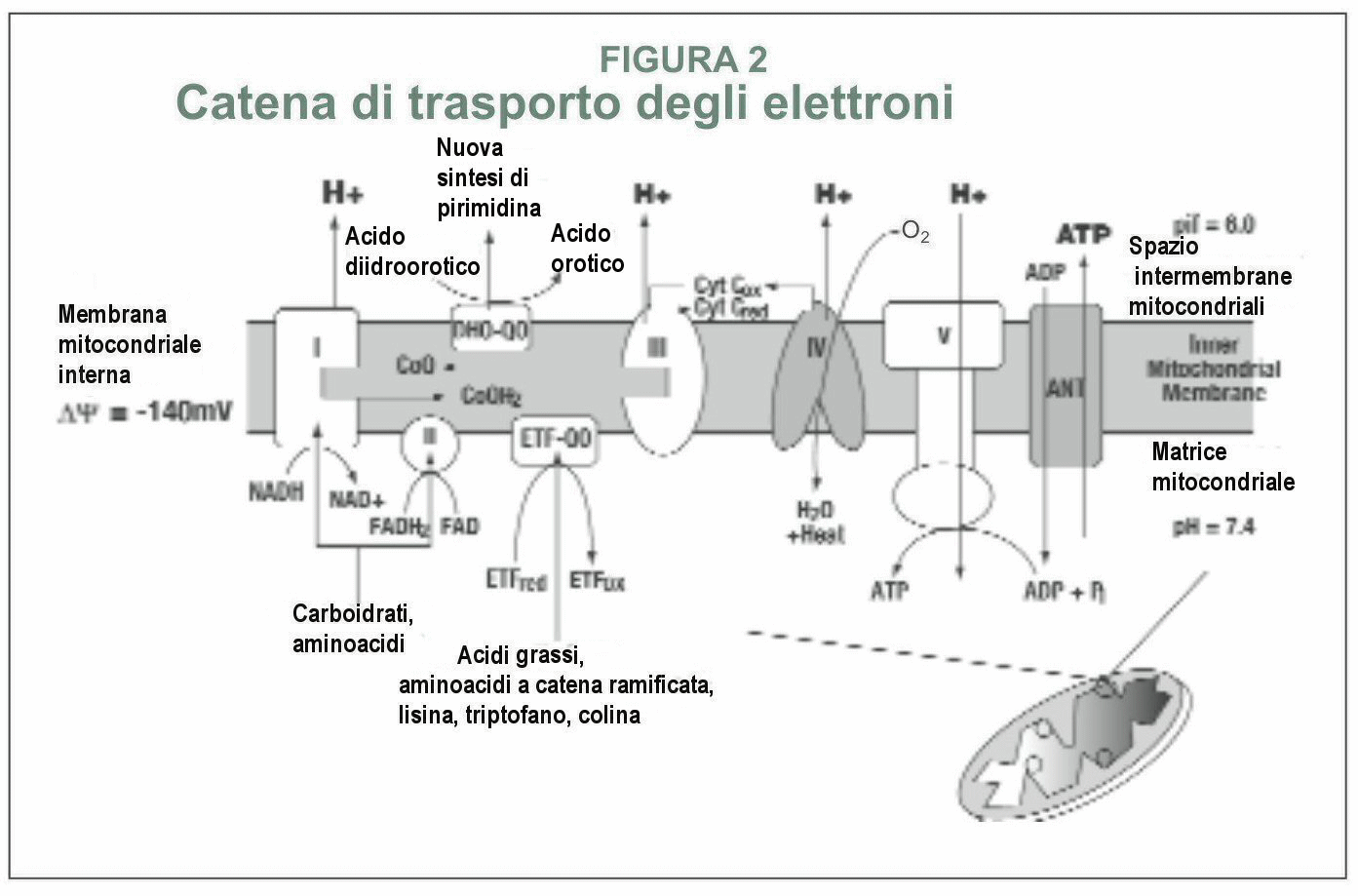
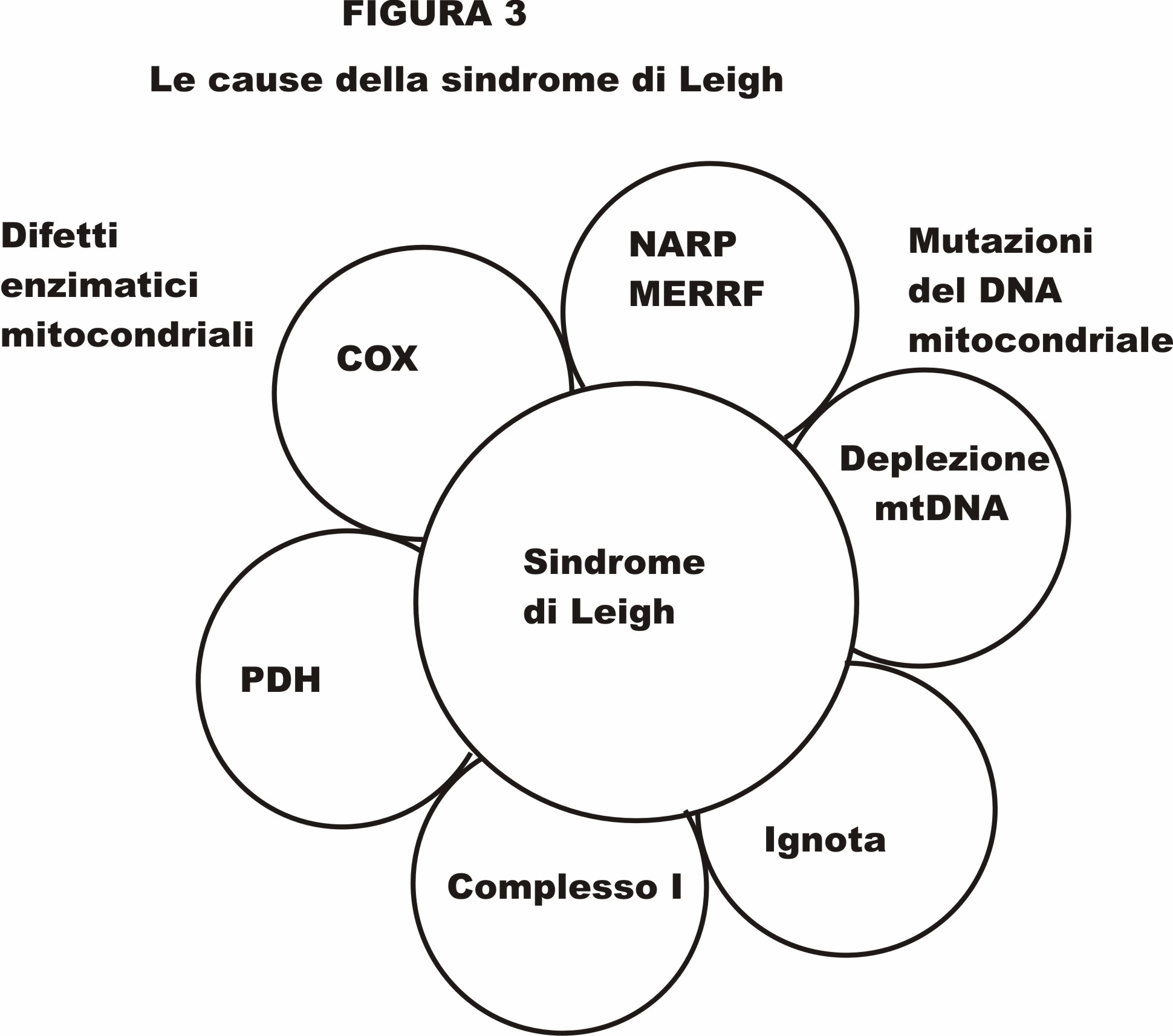
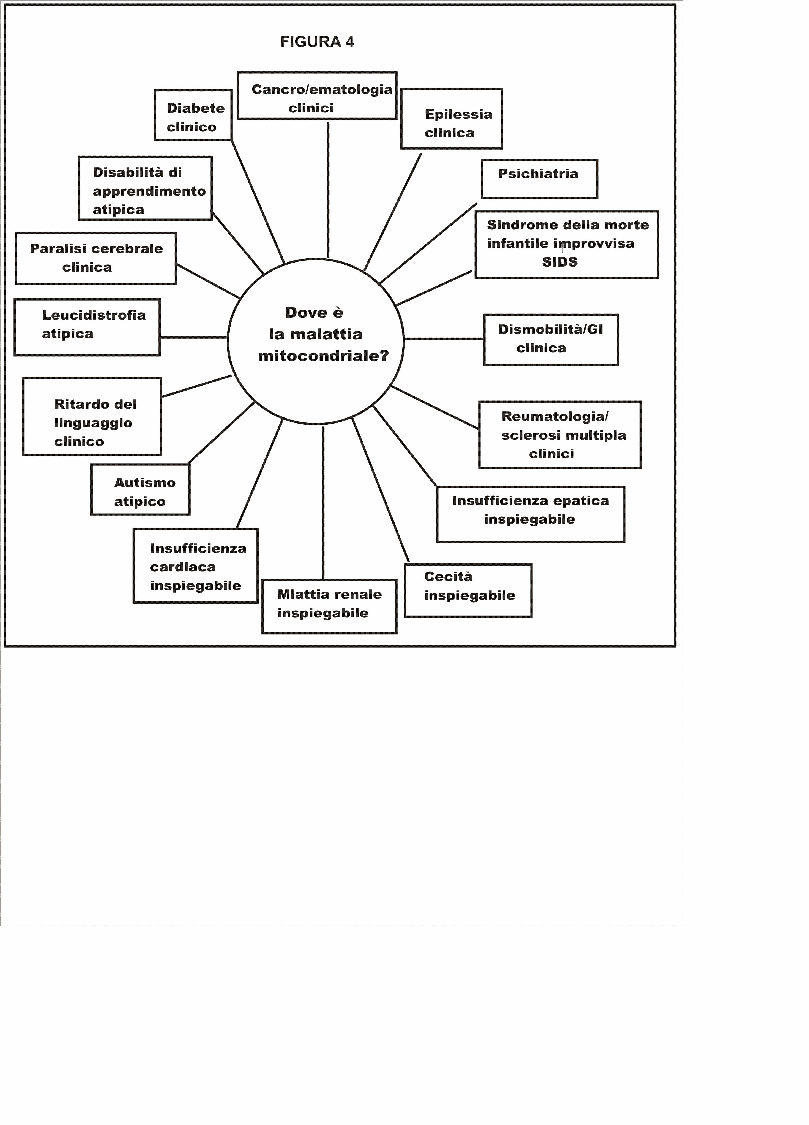
La biologia mitocondriale è una delle aree in più veloce crescita della genetica e della medicina, connessa con discipline scientifiche che vanno dalla embriologia al cancro, alle malattie infettive. Invero, ora si sa che disturbi nel metabolismo mitocondriale giocano un ruolo non solo in rare malattie infantili, ma sono anche coinvolte in molte malattie comuni dell'invecchiamento, incluse malattie del cuore, diabete, malattia di Parkinson e demenza. Questo articolo costituisce un sintetico aggiornamento sulla biologia molecolare per medici, consulenti genetici, ed altri professionisti sanitari che possono trovarsi essi stessi coinvolti nella cura di persone con malattia mitocondriale e loro familiari. Un mitocondrio differente per ogni tessuto- I medici e la maggior parte dei biologi, pensavano che i mitocondri sono piccole salsicce monomorfiche nelle nostre cellule che producono adenosin trifosfato (ATP). In realtà, il mitocondrio assume forme diverse e serve per un gran numero di differenti funzioni metaboliche. La forma di ciascun mitocondrio è caratterizzata dalla cellula specializzata nella quale risiede. (ved. EXCEPTIONAL PARENT, giugno 1977, pp.40-42 per diapositive delle differenti forme di mitocondrio). Tutti dicevano, che ci sono circa 250 differenti tipi di cellule nel corpo umano. I geni espressi in ciascun tipo di cellula sono fatti su misura dalla trascrizione selettiva per far fronte ai bisogni specializzati. Allo stesso modo, ciascun mitocondrio é fatto su misura per far fronte ai bisogni della cellula nella quale risiede. In effetti, ci sono differenti mitocondri con funzioni metaboliche specializzate per molte dei 250 diversi tipi di cellule nel nostro corpo. La maggior parte delle cellule nucleate del nostro corpo contengono da 500 a 2000 mitocondri. Nel cono fotorecettore dell'occhio, i mitocondri costituiscono fino all'80% del volume Intracellularee. Nei muscoli extraoculari come il lateral rectus, essi raggiungono il 60% e nel muscolo cardiaco essi rappresentano il 40% del volume della cellula. Alcuni tipi di cellule hanno solo pochi mitocondri. Le piastrine, per esempio, hanno solo da due a sei mitocondri. I globuli rossi del sangue non contengono mitocondri, ma la loro cellula precursore, il proeritroblasto, é criticamente dipendente dalla funzione mitocondriale fino a che si differenzia in un maturo globulo rosso. I mitocondri sono i soli organelli cellulari che si sapeva avere un loro proprio DNA (DNA mitocondriale o mtDNA), distinto dal DNA nucleare (nDNA). Difetti nel nDNA possono essere ereditati da entrambi i genitori ma, a causa di una peculiarità del processo di fertilizzazione, i difetti dei geni del mtDNA sono ereditati solo dalla madre. Fabbriche energetiche e molto più- La conoscenza comune in biologia e medicina é che la funzione mitocondriale sia solo come "centrali energetiche" per la cellula. Questa eccessiva semplificazione é un errore che ha frenato il nostro progresso verso la comprensione della biologia sottostante la malattia mitocondriale. Servono circa 3000 geni per fare un mitocondrio. Il DNA mitocondriale codifica appena 37 di questi geni; i geni rimanenti sono codificati nel nucleo della cellula e le proteine risultanti sono trasportate al mitocondrio. Solo circa il 3% dei geni necessari per fare un mitocondrio (100 dei 3000) sono destinati alla produzione di ATP. Più del 95% (2900 su 3000) sono coinvolti in altre funzioni legate ai compiti specializzati delle cellule differenziate nei quali risiedono. Questi compiti cambiano mentre noi ci sviluppiamo dall'embrione all'età adulta, ed i nostri tessuti crescono, maturano, e si adattano allo sviluppo postnatale. Queste altre funzioni non collegate dellATP sono intimamente coinvolte con la maggior parte dei più importanti processi metabolici usati da una cellula per costruire, rompere, e riciclare i suoi blocchi di costruzione molecolare. Le cellule non possono perfino fare l'RNA ed il DNA di cui necessitano per svilupparsi e funzionare senza mitocondri. I blocchi costituenti l'RNA ed il DNA sono purine e pirimidine. I mitocondri contengono gli enzimi che controllano la velocità per la biosintesi delle pirimidine (diidroorotato deidrogenasi*) e la sintesi dell'eme (sintetasi dell'acido δaminolevulinico) necessario per produrre l'emoglobina. Nel fegato, i mitocondri sono specializzati alla detossificazione dell'ammoniaca nel ciclo dell'urea. I mitocondri sono inoltre coinvolti nel metabolismo del colesterolo, per la sintesi degli estrogeni e del testosterone, nel metabolismo dei neurotrasmettitori, e nella produzione di radicali liberi e la loro detossificazione. Essi fanno tutto questo in aggiunta alla rottura (ossidazione) dei grassi, delle proteine e carboidrati che noi mangiamo e beviamo.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||