| |||||||||
| |||||||||
QUEST Volume 7, Numero 6, December 2000
Research Updates
Aggiornamenti Di Ricerca
ENZIMA
|
|||||||||


![[QUEST]](http://www.mda.org/images/qpix/q_logoh1.gif)
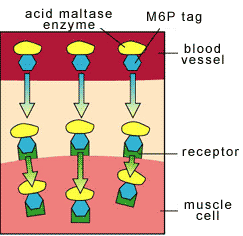 senza mannosio modifiche 6-fosfato senza mannosio Senza modifiche del mannosio 6-fosfato, le molecole acide degli enzimi della
maltasi non possono fornire le
cellule del muscolo dalla circolazione sanguigna. Senza mannosio 6-fosfato tags, acido maltase enzima molecole can't enter Muscoli cellule dalla bloodstream. |
La storia di trattamento di malattia del Pompe va di nuovo ai mid-1990s, dice Chen, quando lui e una squadra che hanno incluso Jer-Yuarn Wu, allora un ricercatore MDA-sostenuto al duca, ha cominciato il lavoro per migliorare sulle strategie più iniziali di trattamento. Due esperimenti in esseri umani per sostituire l'enzima mancante fin dagli anni 70 sono venuto a mancare, Chen spiega, perché i ricercatori non stavano usando il di destra formi dell'enzima.
La Pompe's malattia trattamento story goes back al mid-1990s, says Chen, quando he ed una team che includevano Jer-Yuarn Wu, then un MDA-supportavano investigator a Duke, began work to migliorano on più precoce trattamento strategies. Due esperimenti in umani to replace the mancante enzima come iniziale come the 1970s failed, Chen explains, perché the researchers weren't usando the right forma della enzima.
In un caso, i ricercatori hanno usato una versione della maltasi acida (anche conosciuta come alfa-glucosidasi acida) isolata da un fungo. "che non riusciva,"Chen dice.
In un instance, the researchers usata a version of acido maltase (anche conosciute come acido alfa-glucosidase) isolati dal a fungus. "That era non successful," Chen says.
Un secondo tentativo, questa volta cnella maltasi acida isolata dalle placente
umane, ha avuto più promessa, ma anche venuto a mancare. Chen spiega, "che cosa
è accaduto è che l'enzima placenta-derivato è una forma
matura dell'enzima."
Un secondo attempt, questo tempo con acido maltase isolati dal umana placentas, aveva più promise, ma anche failed. Chen explains, "What happened è che la placenta-derivato enzima è una mature forma della enzima."
Una modifica molecolare ha denominato il mannosio 6-fosfato
"già era stata fenduta fuori,"dice, quando l'enzima chimicamente è stato
cambiato dal relativo acerbo
(precursore) formi al relativo maturo formano. A questo punto, non poteva più da
entrare nelle cellule del muscolo una volta
iniettato nella circolazione sanguigna. È la modifica del mannosio 6-fosfato
che permette che le cellule
del muscolo prendano l'enzima e lo designino come bersaglio allo
scompartimento cellulare in cui fa il relativo lavoro metabolico.
A molecolari tag chiamate mannosio 6-fosfato "aveva already been cleaved off," he says, quando the enzima era Chimicily cambiamentid dal its immature (precursore) forma to its mature forma. A questo Puntiforme, essa non era più lunga able to enter Muscoli cellule quando injected dentro the bloodstream. It's the mannosio 6-fosfato tag che allows Muscoli cellule to take up the enzima e bersglio it al cellulare compartimenti dove it does its metabolica work.
|
con mannosio modifiche 6-fosfato con mannosio 6-fosfato tags Quando le modifiche del mannosio 6-fosfato sono aggiunte alle
molecole acide degli enzimi della maltasi, le molecole attaccano ai ricevitori
(luoghi di aggancio) sulle cellule
del muscolo e sono più profondo trasportato all'interno delle
cellule, dove sono necessari.
Quando mannosio 6-fosfato tags sono aggiunsero ad acido maltase enzima molecole, the molecole stick to recettori (docking siti) on the Muscoli cellule e sono erano portatori deeper interno the cellule, dove they're necessaria. |
Con gli avanzamenti nella tecnologia molecolare, entro gli anni 90 era un aspetto diretto per inserire un gene umano nelle cellule nonhuman e per farlo preparare un enzima voluto per uso negli studi clinici. La squadra del Chen ha scelto fare le cellule cinesi dell'ovaia del criceto fare prendere il gene per la forma del precursore del gene acido umano della maltasi e la maltasi acida cnella modifica del mannosio 6-fosfato.
Con advances in molecolari technology, delle 1990s essa era a straightforward materia to insert a umana gene dentro nonhuman cellule e hanno them make a desired enzima per use in cliniche studi. Chen's team chose to hanno cinesi criceto ovary cellule take up il gene per the precursore forma della umana acido maltase gene e make acido maltase con la mannosio 6-fosfato tag.
Gli studi hanno confermato che l'enzima ha funzionato in cellule umane in un piatto ed allora, in 1998, in quaglie in tensione cnella malattia del Pompe.
Studi confermate che la enzima worked in umana cellule in una dish e then, in 1998, in live quails con Pompe's malattia.
Dopo, i ricercatori hanno regolato su produzione ed hanno provato il trattamento sui bambini. L'annuncio di ottobre, fatto ad una riunione della società americana della genetica umana a Filadelfia, era una radura va segnale per la strategia degli enzimi in esseri umani.
Next, researchers scaled up produzione e tried the trattamento on babies. La October announcement, made a a meeting della American Society of gene umanotica in Philadelphia, era a chiara go segnale per the enzima strategia in umani.
Genzyme e Chen desiderano espandere la prova clinica in bambini e più successivamente in bambini ed in adulti con una forma meno severa della mancanza acida della maltasi. MDA ha offerto di funzionare con loro su una prova clinica espansa di Pompase, l'enzima Chen e colleghe sviluppati ed a quale Genzyme ora tiene i diritti commerciali.
Genzyme e Chen want to expand the cliniche trial in babies e più tardi nei bambini e adulti con una meno grave forma of acido maltase Carenza. MDA ha offered to work con them on un espanse cliniche trial of Pompase, the enzima Chen e colleagues sviluppate e nella quale Genzyme ora Tiene commercial rights.
MDA ha costituito un fondo per la ricerca sia nella terapia del gene che nella terapia del rimontaggio degli enzimi per la mancanza acida della maltasi.
MDA ha funded ricerche in entrambi gene Terapia e terapia sostitutiva enzimatica per acido maltase Carenza.
Allo stesso tempo, una ditta farmaceutica olandese, pharming, ha avuto certo successo con una prova iniziale in infanti usando un enzima che acido della maltasi l'azienda è manufacturing nei conigli che trasportano il gene acido umano della maltasi. E Novazyme, un'azienda nella città di Oklahoma, ha ricevuto la condizione orphan della droga dalla gestione della droga e dell'alimento per lo sviluppo della relativa versione della maltasi acida umana. La condizione orphan della droga è un programma federale che dà ad aziende i motivi finanziari per sviluppare le droghe che non sarebbero normalmente vantaggiose.
A the stessa tempo, a Olandesi pharmaceutical company, pharming, ha aveva alcuni success con un iniziale trial nell'infanzia usando un acido maltase enzima the company è manufacturing in rabbits portatori the umana acido maltase gene. And Novazyme, a company in Oklahoma City, ha ricevuto Orphan Farmaci status dalla Food e farmaci Administration per the sviluppo dei suoi version of umana acido maltase. Orphan Farmaci Status è una federal program che gives companies financial incentives to sviluppo farmaci che normalmente wouldn't be profitable.
MDA GRANTEES FANNO GLI AVANZAMENTI DELLA GENETICA MDA GRANTEES MAKE GENETICS ADVANCES
La società americana della riunione umana della genetica, tenuto a Filadelfia in ottobre, ha montrato più di 100 progetti di ricerca neuromuscolare, molti di cui siano costituiti un fondo per da MDA.
La American Society of gene umanotica meeting, held in Philadelphia in October, showcased più che 100 neuromuscolare ricerche projects, molte della quale erano funded by MDA.
Ad un passo avanti drammatico per il trattamento di una malattia neuromuscolare mortale, Grantee Y di MDA.T. Chen ha annunciato i risultati positivi di una prova di trattamento in infanti cnella mancanza acida della maltasi. (Veda "I Bambini Dei Benefici Di Trattamento enzimatico".)
In una drammatica passo forward per trattamento di una fatale neuromuscolare malattia, MDA grantee Y.T. Chen announced risultati positivi di una trattamento trial nell'infanzia con acido maltase Carenza. (Vedi "Enzima Trattamento Benefits Babies".)
Altri punti culminanti di riuniUna erano:
Altro meeting highlights erano:
Modello del topo per SBMA
Topo Model per SBMA
J.R. Morrison e D.M.. de Krester e colleghe all'università di Monash, L'Australia, hanno sviluppato il primo modello del topo di atrofia muscolare bulbare spinale (malattia del Kennedy o di SBMA). In questa malattia lentamente progressiva, le cellule dicontrollo del nervo sono influenzate.
J.R. Morrison e D.M. de Krester e colleagues a Monash University, Australia, hanno sviluppate il primo topo model of spinali bulbare muscolare atrofia (SBMA o Kennedy's malattia). In questo lentamente progressive malattia, Muscoli-controllanti nervo cellule sono affetti.
Il nuovo modello del topo, quale trasporta la mutazione umana, sembra riprodurre fedelmente i problemi neurologici visti in esseri umani con SBMA e dovrebbe dimostrare un attrezzo utile per lo studio delle cause della malattia.
La new topo model, la quale carries the umanUna mutazione, seems to faithinteramente reproduce the neurologiche problemi visto in umani con SBMA e devono prove a utile tool per investigating the causa della malattia.
I geni che proteggono da SLA hanno identificato nei topi
Genes che Protect Against SLA Identificata in Topo
scienziato MDA-costituito un fondo per C.B. Kunst dell'istituto di Eleanor Roosevelt a Denver ed i colleghe hanno notato che alcune linee dei topo non sviluppano i sintomi della sclerosi di laterale amyotrophic malgrado avere la stessa mutazione genetica che causa una forma ereditata della malattia in esseri umani. Similmente, alcuni esseri umani non sviluppano SLA malgrado avere una mutazione SLA-causante identificata.
MDA-funded scientist C.B. Kunst della Eleanor Roosevelt Institute in Denver e colleagues hanno notati che alcuni linee del topo don't sviluppo sintomi of amyotrophic lateral sclerosis nonostante aventi the stessa genetica mutazione che causa un ereditata forma della malattia in umani. Similarly, alcuni umani don't sviluppo SLA nonostante aventi un identificate SLA-causante mutazione.
Il gruppo ha allevato selettivamente i topo nel tentativo d'identificare un gene specificoo o i geni che trasportano la protezione da SLA. Hanno localizzato gli effetti protettivi alle regioni su tre cromosomi del topo. Una delle regioni contiene un gruppo dei geni pensati per partecipare ad un'altra malattia del neurone motorio, atrofia muscolare spinale. Capendo se, e come, i geni di SMA hanno potuto partecipare a SLA hanno potuto avanzare significativamente la conoscenza di patologia di SLA.
La gruppo bred the topo selettivamente in un attempt to identificarono una specifico gene o geni che convey protection dal SLA. Essi've localizzata protettivi effetti to regioni on tre topo cromosomi. Uno della regioni contiene a gruppo of geni pensarono essere coinvolti in altro neuroni motori malattia, spinali muscolare atrofia. Understanding whether, e how, the SMA geni potesse essere coinvolti in SLA could significativamente advance knowledge of SLA patologia.
DMD Research Progressione
Diagnosi: Come molte come 40 percento of boys con DMD mancanza the grossi "gaps" (delezioni) nei loro distrofina geni che possono essere scoperta con comuni genetica diagnostic tests. infatti, queste boys hanno altre kinds del gene flaws, conosciute come mutazioni puntiformi, la quale sono tiny "spelling errors" nel DNA code o tiny additions o subtractions del DNA che nevertheless render il gene nonfunzionale. (Vedi "Simply Stated," per un spiegazione of differenti kinds del gene flaws.)
Detecting tipo tiny cambiamenti nel molto grossi gene distrofina è una tempo-consuming processo che isn't done routinely come a parti della diagnosi. Now, come un offshoot della MDA-funded trial to esame the Abilità della antibiotic gentamicin to overcome a specifico mutazione puntiforme in DMD, MDA grantee Jerry Mendell of Ohio State University in Columbus e colleagues hanno sviluppate a lunga-necessaria, fast e accurate metodo per risultati queste cambiamenti nel gene distrofina. usando a technique they call DOVAM-S o Ricerca of Virtually Tutti Mutazioni-SSCP, the gruppo era able to identificarono Mutazioni specifiche in più che 90 percento of DMD pazienti who provarono negative per the più comune grossi distrofina delezioni.
Grosse-Scale Cambiamenti: Due MDA-supportavano gruppi Capoed by Louis Kunkel of Bambini's Hospital in Boston e Eric Hoffman of Bambini's National Medical Center in Washington, D.C., hanno usata cutting-edge microarray (computer chip) technology to studio l'attività modelli of migliaia of geni nel Muscoli of people con DMD, arto-girdle MD e congenita MD Muscoli e normale Muscoli. Essi've trovata che, in ogni of queste Malattie, la proteina produzione by numerosi geni è alterato e possono essere misurati.
Con studying Muscoli tessuto sotto differenti circostanze, they speranza per ottenere new insights dentro la malattia processo e potenzialmente discover new molecolari obiettivi per Terapia.
Therapies: On il gene Terapia front, MDA grantee Jeffrey Chamberlain della University of Michigan ha sviluppate una serie of "micro-dystrophins." Queste forme della proteina mancante in DMD sono sottili abbastanza to fit dentro un adeno-associata virus designed to deliver correct geni ai muscoli cellule, mentre retaining the Abilità to correct Patologia muscolare, un minimo al livelli della più leggero Dm di Becker, Chamberlain said.
La britanniche gruppo led by MDA grantee Kay Davies of Oxford University riportati che l'aumento distrofina's sorella molecole Utrofina in tutti tessuti isn't tossici to topo e it Efficacely protegge contro the mancanza of distrofina. La gruppo è still vagliatura per a sottili molecole capaci of l'aumento Utrofina produzione nei muscoli, un approccio che Può result in una farmaci Terapia per DMD.
Nuove Tool per sviluppoing SMA Trattamenti
A recent MDA-funded studio paves the way per farmaci treatments che could compensate per il genetica flaw sottolineare spinali muscolare atrofia (SMA), una malattia che porta a paralisi attraverso le La distruzione dei neuroni motori (nervo cellule che controllo Muscoli).
Niniziale tutti casi delle SMA sono causata da delezione della SMN1 gene, la quale normalmente producente the essenziali sopravvivenza neuroni motori (SMN) proteina. Everyone ha a backup gene SMN chiamate SMN2, ma it producente molto basso livelli of attivi proteina SMN -- solitamente non abbastanza to interamente substitute per the mancante SMN1 produce.
Ma it's possibili to boost produzione di proteina SMN dal SMN2, in accordo al new studio, un international collaboration che includevano MDA grantees Christian Lorson of Arizona State University in Tempe e Elliot Androphy dalla Nuove England Medical Center e Tufts University School of Medicine in Boston. In theory, a alto abbastanza boost to SMN2 poteva essere usata di trattare SMA.
Precedenti ricerche la maggior parterarono che proteina produzione dal SMN2 è estremamente sensibile in una molecolari editing processo che cuts e pastes RNA -- the Chimici intermediate fra DNA (the materiale che makes up geni) e proteina. A causa di una tiny differenza fra the SMN1 e SMN2 geni, che processo spesso clips off a piece of SMN2 RNA che's critica to rendendo interamente funzionale proteina SMN.
La new studio, pubblicati in August nel proceedings della National Academy of Science, mostra che un proteina chiamate Htra-ß1 can attach to SMN2 RNA e protect it dal questo overzealous editing. Quando injected dentro isolati umana cellule, Htra-&sxlig;1 causata the cellule to aumento their livelli of "tutta lunghezza" SMN2 RNA by riguardo threefold.
Sebbene che effetto è incoraggiare, it's probabilmente non grossi abbastanza to consider usando Htra-&sxlig;1 come a trattamento per SMA, Lorson said. infatti, he plans to use Htra-&sxlig;1 "in sviluppano screens per sottili molecole e sottili composizioni che can do the stessa thing" -- solo meglio. Htra-&sxlig;1, he spiegato, will serve come a yardsZecca per measuring gli effetti of centinaia of migliaia of potenziale farmaci che might boost tutta lunghezza SMN2 RNA to clinicamente di beneficio amounts.
In altro sviluppo , the farmaci gabapentin, marketed per seizure Malattie come Neurontin by Pfizer, failed to mostrare ogni beneficio per people con SMA, in accordo to studio team membro Robert Miller, un MDA-supportavano neurologist e cliniche investigator a California Pacific Medical Center in San Francisco.
Gabapentin Parzialely blocchi glutamato, a natural centrale sistema nervoso Chimici che laboratorio evidenza suggerita potesse essere tossici to fragile nervo cellule in SMA.
A separate trial of gabapentin in SLA recentemente failed to mostrare ogni beneficio in che malattia.
Risultati di una un-anno, MDA-supportavano studio of 64 people con SMA tipi 2 e 3 ( meno grave che tipo 1) non mostrava differenze in Risultano fra those dati gabapentin e those receiving a placebo.
Le risultanze erano presentarono Oct. 17 durante a meeting della American Neurologico Associazione in Boston. basati on queste ritrovamenti, the use of gabapentin in SMA cannot be recommended, say MDA advisers.
Nuove Insights dentro malattie mitocondriali
Nuove visioni delle malattie mitocondriali
Tre studi recenti hanno portato gli scienziati più vicino ai
trattamenti di sviluppo per le malattie causate dai difetti in
mitocondri, le fabbriche vitali di produttore d'energia hanno trovato nella
maggior parte delle cellule.
Tre recent studi hanno brought scientists closer to sviluppano treatments per malattie causata da difetti in mitocondri, the vital energia-producente factories trovata in maggior parte cellule.
Come molte malattie, le malattie mitocondriali sono causate dalle mutazioni (cambiamenti) in DNA (il prodotto chimico che compone i nostri geni). Ma soltanto alcune malattie mitocondriali sono causate dalle mutazioni nel capito in il più bene, la maggior parte del tipo abbondante di DNA -- il genere ha imballato nel centro di controllo delle cellule conosciuto come il nucleo. Altre malattie mitocondriali sono causate dalle mutazioni in DNA alloggiato nel DNA dei mitocondri essi stessi -- che è usato per fare le proteine mitocondriali essenziali.
Like molte malattie, malattie mitocondriali sono causata da mutazioni (cambiamenti) in DNA (the Chimici che makes up our geni). Ma solo alcuni malattie mitocondriali sono causata da mutazioni nel meglio compresa, maggior parte abbondanti tipo del DNA -- the kind packed dentro the cellule controllo center conosciute come the nucleo. Altro malattie mitocondriali sono causata da mutazioni in DNA housed nel mitocondri themselves -- DNA che's usata per fare essenziali mitocondriale proteine.
In due studi indipendenti, gli scienziati hanno prodotto i primi topo che hanno malattie mitocondriali causate dalle mutazioni di DNA mitocondriale (mtDNA). Anche se né l'unoo né l'altro sforzo del topo ha le caratteristiche esatte di una malattia mitocondriale umana particolare, saranno utili per progettare i trattamenti che neutralizzano gli effetti offensivi delle mutazioni di mtDNA.
In due indipendente studi, scientists hanno prodotto il primo topo che hanno malattie mitocondriali causata da mutazioni del DNA mitocondriale (mtDNA). Sebbene neither topo strain ha the esatta caratteristiche di una particolare umana Malattia mitocondriale, they'll be utile per designing treatments che counteract the damaging effetti delle mutazioni del mtDNA.
Uno sforzo del topo è stato generato dagli scienziati all'università di Emory a Atlanta, chi ha presentato il loro lavoro alla società americana per la riunione umana della genetica in ottobre. L'altro sforzo del topo è stato generato da un gruppo degli scienziati giapponesi, chi ha pubblicato il loro lavoro nell'emissione di ottobre della genetica della natura. Questo sforzo ha lo stesso tipo di mutazione di mtDNA che causa la sindrome mitocondriale umana di Kearns-Sayre di malattie (KSS) ed il oftalmoplegia esterna progressiva (PEO).
Uno topo strain era creato by scientists a Emory University in Atlanta, la quale presentava their work a the American Society per gene umanotica meeting in October. Gli altri topo strain era creato da una gruppo of giapponese scientists, who pubblicati their work nel October issue of Nature Genetica. Questa strain ha the stessa tipo of mutazione del mtDNA che causa the umana malattie mitocondriali sindrome di Kearns-Sayre (KSS) e oftalmoplegia esterna progressiva (PEO).
KSS e PEO sono causati dalle mutazioni di mtDNA che eliminano un tipo
speciale di RNA mitocondriale (l'intermediario del prodotto chimico
fra DNA e proteina). Questo RNA -- tRNA denominato -- permette ai mitocondri di
produrre le
loro proprie proteine; quando manca, tutte quelle proteine essenziali mancano,
anche.
KSS e PEO sono causata da mutazioni del mtDNA che eliminate a special tipo of mitocondriale RNA (the Chimici intermediate fra DNA e proteina). Questa RNA -- chiamate tRNA -- enables mitocondri per produrre their own proteine; quando it's mancante, tutti of those essenziali proteine sono mancante, too.
Uno studio da una squadra internazionale di scienziati suggerisce le
malattie mitocondriali come KSS e PEO potrebbero essere trattati
sostituendo il tRNA mitocondriale mancante con il tRNA prodotto nel
nucleo (che funziona normalmente in parti dei mitocondri esterni delle
cellule). Nello studio, pubblicato nell'emissione di settembre di scienza, i
ricercatori hanno identificato i segnali che promuovono
l'importazione dei tRNAs nucleari nei mitocondri, ed indicato che quei tRNAs
nucleari potrebbero produrre le proteine
mitocondriali.
A studio by un international team of scientists suggerisce malattie mitocondriali simili KSS e PEO poteva essere trattata by replacing the mancante mitocondriale tRNA con tRNA prodotto nel nucleo (la quale normalmente funzioni in parti della cellule sul lato esterno mitocondri). Nella studio, pubblicati nel September issue of Science, the researchers identificate segnali che promuovano the import of nucleari tRNAs dentro mitocondri, e mostrarono che those nucleari tRNAs could manufacture mitocondriale proteine.
Difficoltà fisica e mentale di affaticamento quelli con magnesio
Fisico e Mental Fatica Trouble Queste Con MG
Ventotto genti con i gravis e 34 di miastenia senza alcuna malattia neuromuscolare (un gruppo di controllo) sono state studiate per sondare il ruolo di affaticamento in magnesio. Jonathan Goldstein, un neurologo che dirige la clinica di MDA all'università di Yale a Nuove Haven, Connett., e James Gilchrist, un neurologo che dirige la clinica di MDA all'ospedale dell'Rhode-isola nel providence, erano sul gruppo di studio, quale ha pubblicato i relativi risultati nell'emissione di settembre del muscolo e del nervo.
Twenty-otto people con miastenia grave e 34 senza ogni neuromuscolare malattia (un gruppo di controllo) erano studiarono to probe il ruolo of fatica nella MG. Jonathan Goldstein, a neurologist who directs the MDA clinic a Yale University in Nuove Haven, Conn., e James Gilchrist, a neurologist directing the MDA clinic a Rhode Island Hospital in Providence, erano on the studio team, la quale pubblicati its ritrovamenti nel September issue di muscolo e Nervi.
Nella forma più comune di magnesio, il sistema immune attaca i ricevitori dell'acetilcolina, strutture sulle cellule del muscolo che ricevono i segnali dai nervi. La debolezza oscillante di cause immunologiche di attacco, descritto spesso dai pazienti come affaticamento. L'affaticamento fisico come conseguenza della debolezza del muscolo è previsto nel disordine, i ricercatori dicono, ma sono stati sorpresi imparare che l'affaticamento (pens-relativo) conoscitivo inoltre si presenta spesso in magnesio.
Nella maggior parte comuni forma of MG, the immune sistema attacchi the acetilcolina recettori, strutture sui muscoli cellule che receive segnali dal nervi. La immunologic attacchi causa fluctuating debolezza, spesso descritti by pazienti come fatica. Fisico fatica come risultato di debolezza muscolare è aspetti nel malattia, the researchers say, ma essi erano surprised to learn che cognitive (thinking-relativa) fatica anche si ha spesso nella MG.
I ricercatori hanno trovato che quelli con magnesio hanno segnalato sensibilmente più di affaticamento fisico e conoscitivo che quelli nel gruppo di controllo e che entrambi i generi di affaticamento hanno interessato il loro mentale, funzionamento fisico e sociale.
La investigators trovata che those con MG riportato significativamente più fisica e cognitive fatica che those nel controllo gruppo e che entrambi kinds of fatica affetti their mentale, fisica e social functioning.
I ricercatori dicono che non possono non vedere motivo libero per l'affaticamento conoscitivo in magnesio, poiché il cervello non si pensa per partecipare alla malattia. Tuttavia, notano che l'affaticamento fisico può influenzare la percezione della persona del suo livello del tiredness mentale; e, notano, il apnea di sonno (cessazione periodica di respirazione durante il sonno) a causa della debolezza del muscolo potrebbe causare l'affaticamento conoscitivo durante il giorno.
La researchers say they can vedi no chiara reason per the cognitive fatica nella MG, sino the encefalo isn't pensarono essere coinvolti nel malattia. Comunque, they note che fisica fatica Può influenzano a person's percezione of his livelli of mentale tiredness; e, they note, Apnea nel sonno (periodic cessation of breathing durante sonno) perché di debolezza muscolare could cause cognitive fatica durante the giorno.
Soggetto di effetti secondari del prednisone dello studio a lungo termine
Prednisone Effetti collaterali Topic of lunga-Term Study
Il prednisone della droga del corticosteroide è stato trovato per ritardare il deterioramento del muscolo in ragazzi cnella distrofia muscolare di Duchenne (DMD), ma con molti effetti secondari.
La corticosteroid farmaci prednisone ha been trovata to lenta Muscoli deteriorazione in boys con distrofia muscolare di Duchennee (DMD), ma con molte effetti collaterali.
Gli studi sono stati intrapresi dai medici a quattro cliniche di MDA: l'università di Rochester (N.Y.) Centro Medico; L'Ohio Dichiara gli ospedali dell'università a Columbus; Centro universitario medico di Vanderbilt a Nashville, Tenn.; e centro universitario medico de Washington in st. Louis. I risultati sono stati uniti con quelli dei ricercatori che vedono i pazienti attraverso le cliniche connesse con l'università di Alberta a Edmonton, Il Canada.
Lo studio era conducted by medici a quattro MDA cliniche: the University of Rochester (N.Y.) Medical Center; Ohio State University Hospitals in Columbus; Vanderbilt University Medical Center in Nashville, Tenn.; e Washington University Medical Center in St. Louis. Risultati erano combinata con those of investigators seeing pazienti attraverso le cliniche associata con la University of Alberta in Edmonton, Canada.
I ricercatori hanno documentato gli effetti secondari prednisone-collegati in ragazzi su DMD che ha cominciato sulla droga fra 1986 e 1989. I dati sono stati raccolti in 1991, 1995 e 1999. Fra 1991 e 1999, il numero di pazienti che sono seguiti ribassanti da 226 a 100, con 51 tranquilli su prednisone.
La investigators documentarono prednisone-associata effetti collaterali in boys con DMD who iniziavano on the farmaci fra 1986 e 1989. Data erano raccolsero in 1991, 1995 e 1999. fra 1991 e 1999, the numero dei pazienti essendo seguita declined dal 226 to 100, con 51 still on prednisone.
La loro età media in 1999 era di 18 anni, ed il tempo medio su prednisUna era di sette - 12 anni.
Their media età in 1999 era 18 anni, e the media tempo on prednisUna era seven to 12 anni.
Di quei pazienti o genitori che hanno scelto interrompere il prednisone o riducono il dosaggio, 85 per cento hanno citato il guadagno inaccettabile del peso come il motivo per fare così. Circa 13 per cento hanno dato le oscillazioni di umore o altri problemi del comportamento come il motivo. Altre complicazioni che si sono presentate durante il periodo lungo di studio hanno incluso le cataratte in 19 ragazzi; vertebre fratturate in quattro ragazzi; fratture delle ossa polmonehe insieme con le cadute in 21 ragazzo; alta pressione sanguigna in tre ragazzi; diabete in un ragazzo; ed altezza corta con maturazione sessuale in ritardo in sette ragazzi.
Of those pazienti o genitori who chose to discontinue prednisone o ridurre the dosaggio, 85 percento citavano unacceptable aumento di peso come the reason per doing so. Alcune 13 percento gave mood swings o altre comportamentoal problemi come the reason. Altro complicazioni che avvenuta durante the lunga studio periodo includevano cataratta in 19 boys; fractured vertebrae in quattro boys; fratture of lunga bones in congiunzione con cadute in 21 boys; alto pressione sanguigna in tre boys; diabete in un ragazzo; e bassa statura con ritardata sexual maturazione in seven boys.
I ricercatori suggeriscono che l'amministrazione migliorata di guadagno eccessivo del peso permetterebbe a tantissimi pazienti di trarre beneficio dal trattamento di lunga durata con prednisone.
La researchers suggeriscono che migliorato gestione of eccessiva aumento di peso would permit a grandi numero dei pazienti to beneficio dal lunga-term trattamento con prednisone.
I risultati sono stati presentati ad una riunione dell'associazione neurologica americana a Boston in ottobre.
La Risultano erano presentarono a a meeting della American Neurologico Associazione in Boston in October.
Ancora un'altra Proteina, Gene implicato in LGMD
Yet Un altro Proteina, Gene Implicated in LGMD
Il gene per il miotilina, una proteina situata nella parte della cellula del muscolo che permette che si contragga e si distenda, è stato confermato per svolgere un ruolo nel causare membro-cingono il tipo muscolare 1A di distrofia. Grantee Marcy Speer di MDA al centro medico della Duke University in Durham, N.C., era sulla squadra che ha segnato il gene con esattezza di miotilina come essendo responsabile di LGMD in una grande famiglia nordamericana della discesa tedesca. L'individuazione è pubblicata in settembre. 1 emissione della genetica molecolare umana.
il gene per miotilina, una proteina localizzato nel parti della Muscoli cellule che allows it to contract e relax, ha been confermate to giocare un ruolo nel causare arto-girdle distrofia muscolare tipo 1A. MDA grantee Marcy Speer a Duke University Medical Center in Durham, N.C., era on the team che pinPuntiformeed the miotilina gene come essendo responsabile per LGMD in una grossi nordamericani famiglia of tedesche discendenti. Il ritrovamento è pubblicati nel Sept. 1 issue of Umano molecolare Genetica.
Almeno i geni differenti un dozzina inscatolano ciascuno, una volta difettoso, conduca a LGMD, ora riconosciuto come un gruppo dei disturbi che interessano principalmente i muscoli della spalla e delle cinture pelviche, quale stabilizzano l'armi ed i piedini superiori.
A least a dozzine differenti geni can ogni, quando difettose, lead to LGMD, ora identificata come a gruppo of Malattie che colpisce maggiorpartemente the Muscoli della spalla e pelvic girdles, la quale stabilizzare the superiore braccia e gambe. ![]()
|
Ritorno a Fonama.org Home Page |
Alla pagina originale
|
|