| ||||||||
|
| ||||||||
QUEST | Fascicolo corrente | Numeri precedenti | Storie per argomento | Storie dalla ricerca | Subscribe | Advertise | Contents of This Issue
AGGIORNAMENTI DALLA RICERCA
PROGRESSI NELLA MIOPATIA MITOCONDRIALECosa accade quando le cellule esauriscono l'energia In quasi tutte le cellule del corpo ci sono delle piccole centrali dette mitocondri. La teoria vuole che 1,5 miliardi di anni fa, i mitocondri fossero cellule indipendenti, probabilmente dei batteri, e che siano stati inglobati in cellule di organismi superiori che necessitavano di una fonte rapida di energia. Quello che i mitocondri fanno per la cellula -- rapida produzione di energia, lo spiega un ricercatore della MDA Douglas Wallace della Emory University di Atlanta, dove è rimasto per studiarlo per più di dieci anni. I mitocondri hanno il loro proprio DNA (geni), ma le loro funzioni sono governate anche dai geni del nucleo cellulare, il centro principale di comando. I mitocondri operano come sotto un sistema di doppio controllo.
I disturbi mitocondriali, incluse le miopatie mitocondriali (disturbi muscolari derivanti da malfunzioni mitocondriali), possono essere la conseguenza di anomalie del DNA dei mitocondri o del DNA del nucleo della cellula che altera i mitocondri. Ora, sono stati sviluppati due modelli di miopatia mitocondriale nei topi, come punto di partenza per aiutare gli scienziati a risolvere alcuni dei misteri di questo tipo di disturbo. Nel primo modello, Wallace ed i suoi colleghi alla Emory incrociano topi senza geni funzionanti per la proteina conosciuta come ANT1. (Il gene per l'ANT1 è nel nucleo della cellula.) Questa proteina normalmente trasporta l'ATP dai mitocondri nel corpo principale della cellula (citoplasma ndt). Senza questa, l'ATP prodotto si accumula nei mitocondri e non può soddisfare le necessità energetiche della cellula. I topi con carenza di ANT1 hanno cuore ingrandito, muscoli con "ragged red fiber", segno indicativo di mitocondri anormali nell'uomo, e non tollerano l'esercizio fisico -- stessi sintomi che si vedono negli esseri umani con disturbi mitocondriali. Wallace nota che non sono stati trovati umani con carenza di ANT1 ma, dice, non c'è un'unica regola come in alcune miopatie mitocondriali che possa causare questo difetto. Wallace dice che il modello con carenza di ANT1 è la prima chiara dimostrazione della relazione di causa-effetto fra il limitato apporto di ATP (la molecola energetica) e l'effetto fisiologico visto nelle miopatie mitocondriali. Nel frattempo, un gruppo di ricercatori alla University dell'Alabama a Birmingham e alla University dello Iowa hanno sviluppato un altro modello di miopatia mitocondriale dei topi ed hanno sperimentato una strategia di terapia genetica su questi topi. Questi ricercatori hanno usato un topo con una naturale mutazione genetica in un gene per un enzime mitocondriale conosciuto come SCAD. Il gene è nel nucleo cellulare. Difetti nel gene SCAD sono stati riscontrati in bambini e causano varie anomalie metaboliche muscolari. (L'enzima SCAD si trova normalmente nel fegato, nei reni, nei muscoli scheletrici e nel cuore). I ricercatori incrociarono i topi fino ad avere roditori completamente mancanti di questo cruciale enzima mitocondriale, che normalmente contribuisce alla produzione di ATP. Fecero un ulteriore passo in avanti. Cercarono di vedere quale SCAD poteva rimpiazzare e diventare una cura per disturbi mitocondriali. Usando l'ingegneria transgenica, gli investigatori aggiunsero il gene per SCAD e poterono correggere il deficit di energia nei tessuti dei topi. I ricercatori suggerirono che la terapia genetica con il gene SCAD, fosse diretta solo al fegato (un organo che è facile da investigare e che è vitale nelle affezioni con carenza di SCAD), che dovrebbe essere investigato per le possibilità di trattamento. La mappa della carenza di ANT1 è nel fascicolo di luglio di Nature Genetics, e lo studio sui topi con carenza di SCAD è nel fascicolo di settembre di Human Molecular Genetics. UNA PROTEINA RAGGRUPPA GLI INDIZI PER ATASSIA, E SBMA"Questa è una cosa grandiosa," David Housman, un professore di biologia al Massachusetts Institute of Technology a Cambridge riportato dal The New York Times dell'8 agosto. "Abbiamo girato l'angolo dal guardare i geni a dove noi possiamo cominciare a sviluppare vere sperimentazioni per farmaci". Stava parlando della malattia di Huntington, un disturbo neurodegenerativo del cervello; ma intende che praticamente la stessa cosa può essere detta riguardo ad altre numerose malattie, incluse la Atrofia Muscolare del Bulbo Spinal (malattia di Kennedy) e molte delle atassie spinocerebrali autosomali dominanti (come minimo SCA di tipo 1, 2, 3, 6 e 7). Che cosa hanno in comune delle malattie apparentemente diverse? La loro causa genetica -- una sezione espansa del DNA contenente troppe sequenze ripetitive degli elementi chimici citosina, adenina e guanina, o CAG. Già da quando venne notata la prima volta all'inizio del 1990, l'elenco delle malattie della cosidetta "tripletta ripetuta" è cresciuto enormemente. La ripetizione del CAG è di gran lunga la meglio conosciuta in questo gruppo. Il DNA è il codice, o la ricetta, per la produzione di proteine. CAG è il codice per la glutammina, un componente di alcune proteine. Con troppi codici CAG si avranno troppe molecole di glutammina. Ciò che eccitava i ricercatori per la malattia di Huntington era il trovare che un eccesso di molecole di glutammina in una proteina causava l'accumulo della proteina nel nucleo cellulare, con l'eventualità di portare a morte la cellula. C'era una proteina anormale "raggrumata come un'enorme palla di sudiciume dentro il nucleo," dichiarazione giurata di Stephen Davies, un professore di anatomia allo University College a Londra in Inghilterra, scrive il giornalista del Times riguardo le scoperte sulla malattia di Huntington, che il suo gruppo riportava nel fascicolo dell'8 agosto sulla cellula. Il Dr. Kenneth Fischbeck, professore di neurologia alla University di Pennsylvania e contrattista da molto tempo della MDA, è anche interessato alle ripetizioni di CAG ed alle malattie associate con queste. E' stato il suo gruppo ad identificare per primo le ripetizioni del CAG come causa dello SBMA. Questa estate, lui era fra i ricercatori che trovarono cellule cerebrali con ammassi di proteine nella atassia cerebrale di tipo 3. Il suo gruppo lo ha pubblicato nel fascicolo di agosto sui neuroni. Fischbeck crede che la causa di tutte le malattie da ripetizione delle proteine del CAG, a livello delle proteine e del DNA, sia la stessa, sebbena la proteina colpita in ogni malattia sia diversa, e che le cellule colpite possano essere diverse. Queste differenze rendono la variabilità delle caratteristiche in ogni condizione neurodegenerativa. "La scoperta è l'esistenza di un meccanismo comune per queste malattie," dice Fischbeck, "basato sull'aggregazione di proteine (incollamento) e la tossicità ai neuroni. Ogni nuova scoperta dentro il meccanismo di una malattia ci porta a chiudere su un effettivo e razionale trattamento per questa malattia." Dice il meccanismo appare essere simile alla "malattia poliglutammine". "L'espandersi delle ripetizioni di poli-glutammine cause le aggregazione delle proteine, e questo interferisce presumibilmente con alcuni processi cellulari, come per l'espressione del gene o dei processi RNA, ed eventualmente conduce alla morte della cellula." (L'RNA è il passo chimico fra il DNA e la proteina.) Sounds è arcigno, ma Fischbeck è ottimista. "Per me, la cosa più eccitante riguardo le scoperte recenti è che indicano possibili interventi terapeutici. Le aggregazioni di proteine sembrano precipitare per proteolisi (digestione delle proteine). Se è così, potrebbe essere possibile concepire un trattamento per bloccare la proteolisi o altrimenti interferire con le aggregazioni di proteine. Disponiamo di culture cellulari e di modelli animali possiamo ora sviluppare prove di trattamento. E se il trattamento funziona nelle culture cellulari e negli animali, avremo poi una buona possibilità di lavorare con i pazienti. perché per la suddivisione del lavoro, i trattamenti effettivi per ogni tipo di queste malattie sarà lavoro di altri." TRATTAMENTO DELLA SINDROME MIASTENICA DI LAMBERT-EATONNella sindrome miastenica di Lambert-Eaton (LEMS), la debolezza fluttuante accade perché il sistema immunitario produce anticorpi contro le terminazioni nervose che rilasciano un trasmettitore chimico, l'acetilcolina. Uno studio fatto alla Università dell'Alabama a Birmingham a al Veterans Affairs Medical Center dice che un farmaco conosciuto come guanidine, in combinazione con piridostigmina (Mestinon), è utile e non troppo tossico nella LEMS. Prima molti dottori, pensavano che la guanidina fosse troppo tossica per un uso di routine, aumenta la secrezione dell'acetilcolina da parte della terminazione nervosa. La piridostigmina diminuisce la degradazione dell'acetilcolina così che ce ne sia di più a disposizione per la stimolazione del muscolo. Lo studio è nel fascicolo del mese di settembre di Muscle & Nerve. QUATTRO FARMACI ASSOCIATI OSTEOPOROSIS, PIU' ETIDRONATOI corticosteroidi, farmaci della famiglia del prednisone, sono usati spesso per il trattamento di malattie autoimmunitarie come polimiosite, dermatomiosite, miastenia grave e la sindrome miastenica Lambert-Eaton. Sono usati qualche volta anche nella distrofia muscolare di Duchenne. Questi farmaci, mentre sono qualche volta molto efficaci, hanno però seri effetti collaterali, uno dei più importanti è l'osteoporosi -- perdita di ossa, importante attraverso il decremento del calcio assorbito dalle persone dal cibo o da supplementi. Uno studio canadese nel fascicolo 7 di agosto del New England Journal of Medicine riporta che la combinazione di calcio supplementare con il farmaco etidronato (Didronel) somministrati a rotazione riduceve la perdita di osso e le fratture in persone che prendono i farmaci come il prednisone meglio di quanto faceva la terapia con il calcio da solo. Il calcio è un componente normale dell'osso, e l'etidronato lo attacca all'osso e stabilizza l'osso stesso. PROVA DI NUOVI FARMACI NELLA SMAUn gruppo di ricerca sostenuto dalla MDA alla California Pacific Medical Center di San Francisco, diretto per la MDA dal direttore di clinica Dr. Robert Miller, sta esaminando il farmaco gabapentina (Neurontin) nel trattamento della atrofia muscolare spinale di tipo 2 e 3. Il farmaco è sperimentato anche in altri nove centri nord americani. Persone che si qualifichino per la sperimentazione del farmaco necessitano della conferma della diagnosi di SMA con il test genetico e devono avere una funzione respiratoria adeguata (determinata con un esame conosciuto come capacità vitale forzata). La prova durerà un anno. Alcuni partecipanti riceveranno il gabapentin, che sembra interferire con la formazione della sostanza naturale glutamato, e altri riceveranno un placebo (una sostanza inerte). Il gabapentin è già sul mercato per malattie da carenza ed è provato al California Pacific ed altrove per l'uso nella sclerosi laterale amiotrofica. Il glutamato è tossico alle cellule nervose se ce nè troppo, o una cellula immatura o danneggiata non potesse elaborarlo appropriatamente -- come nel caso della SMA. Per ulteriori informazione sulle prove di San Francisco, chiamare Giovanna Kushner al (415) 923-3604. Per informazioni sui centri di prova, chiama MDA at (800) 572-1717, o visita il sito web MDA al MDAUSA.ORG QUEST | Fascicolo corrente | Numeri precedenti | Storie per argomento | Storie dalla ricerca | Abbonamenti | Inserzioni
| ||||||||


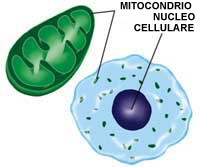 Circa il 90 percento dell'energia necessaria ai tessuti corporei è
prodotta dai mitocondri, che la immagazzinano in una molecola
conosciuta come ATP, il risultato finale di una lunga catena di eventi
chimici. L'ATP deve poi essere trasferita fuori dal mitocondrio nella
parte principale della cellula (citoplasma ndt).
Circa il 90 percento dell'energia necessaria ai tessuti corporei è
prodotta dai mitocondri, che la immagazzinano in una molecola
conosciuta come ATP, il risultato finale di una lunga catena di eventi
chimici. L'ATP deve poi essere trasferita fuori dal mitocondrio nella
parte principale della cellula (citoplasma ndt).