|
Aggiornato nel
settembre 2008
Per la
versione in spagnolo di
questa pubblicazione, clicca
qui:
En Español
|
 |
|
La famiglia Kelly
al Labor Day Telethon della MDA. |
Se state leggendo
questo opuscolo, è probabilmente perché
avete appena ricevuto una diagnosi molto
sconcertante:
miopatia mitocondriale. Cos'è la
miopatia mitocondriale e cosa significa?
Queste sono domande sulle quali ci
dibattemmo mia moglie Jennifer ed io,
quando a nostro figlio Michael venne
diagnosticata una miopatia mitocondriale
nel 1993.
Le miopatie
mitocondriali hanno molte facce
differenti. Come leggerai in questo
opuscolo, sono state identificate
dozzine di tipi di malattie
mitocondriali, con un complesso spettro
di sintomi. Alcuni sintomi possono
essere così lievi da essere
difficilmente notati, mentre altri
possono richiedere un trattamento a
vita.
A Michael la miopatia
mitocondriale causa debolezza muscolare,
crampi muscolari, affaticamento, perdita
dei resistenza e scarso equilibrio. Tu o
il tuo familiare potreste avere sintomi
simili, sebbene ognuno sia un caso
unico.
Quando iniziammo a
capire che Michael aveva una miopatia
mitocondriale, ne fummo naturalmente
molto spaventati e sconcertati per il
futuro. Ma con il passare del tempo
abbiamo imparato che potevamo fare delle
cose che non credevamo nemmeno possibili
- potevamo abituarci all'incertezza,
controllare la paura, far fronte ai
cambiamenti necessari e mantenere
contemporaneamente una vita familiare
"normale" e felice.
Nel 2004 le nostre
vite subirono un nuovo duro colpo,
quando mia moglie, Jennifer, scoprì che
aveva una malattia mitocondriale. I
sintomi di Jennifer sono più gravi di
quelli di Michael; lei ha grave
debolezza muscolare, affaticabilità,
problemi gastrointestinali, problemi
respiratori e difficoltà di
deglutizione.
Questo fascicolo è
stato preparato per aiutarvi a capire le
cause delle miopatie mitocondriali ed i
loro trattamenti. Ci abbiamo trovato
informazioni che sono strumenti vitali
nella gestione delle malattie di Michael
e di Jennifer e per conseguire il
migliore risultato possibile.
Da questo fascicolo
imparerete anche alcune cose
incoraggianti. Per esempio, sebbene
queste siano malattie molto rare, molti
dei loro sintomi sono comuni nella
popolazione generale, come i problemi
cardiaci, gli attacchi epilettici ed il
diabete. Inoltre, sono disponibili buoni
trattamenti medici per aiutare nella
gestione di molti sintomi.
Ricordati sempre che
la ricerca sta continuamente portando
avanti migliori trattamenti, e infine cure per le malattie
mitocondriali. In aggiunta, le persone
con disabilità hanno le maggiori
opportunità come mai prima di utilizzare
la maggior parte delle loro capacità,
come pure per i diritti legali a eguali
opportunità di impiego e di accesso ai
luoghi pubblici. Ai bambini con
disabilità fisiche e cognitive viene
garantita per legge una istruzione
pubblica con qualsiasi supporto
necessiti loro.
La MDA è stata una
alleata molto preziosa dalla quale
continuiamo ad imparare a vivere con la
malattia mitocondriale. “La
MDA è qui per aiutarti,” ti darà
molte più informazioni sui molti servizi
della MDA.
Per noi, Michael non è
la vittima di una malattia o di una
sindrome, ma un giovanotto felice,
affettuoso, del quale noi siamo molto
orgogliosi. Abbiamo constatato che
nessuno può prevedere esattamente come
progrediranno i casi di Michael o di
Jennifer. Siamo stati rallegrati dal
vedere Michael condurre una vita
normale, conquistarsi la sua Boy Scout Eagle Award e venire accolto nella
National Honor Society. Michael ha
mostrato che avere una malattia
mitocondriale non significa
necessariamente rinunciare a portare a
termine gli obiettivi che ti sei posto.
Dalla diagnosi di
Jennifer noi lottiamo per adattarci ai
cambiamenti nelle nostre vite ed è
rassicurante sapere che la MDA è qui per
noi, assistendoci con attrezzature,
cliniche, ricerca continua, ed è proprio
una voce amica che comprende ciò che
stiamo passando.
Mentre tu fronteggi le
sfide che hai davanti, noi ti auguriamo
ogni bene e ti diamo il conforto di
sapere che non sei solo.
Richard
Kelly
Mansfield, Mass.
Nello stesso modo in
cui molte malattie prendono il nome
della parte del corpo che colpiscono
(come le cardiopatie), le malattie
mitocondriali sono così chiamate perché
colpiscono una parte specifica delle
cellule di cui è costituito il
nostro corpo. Specificatamente, le
malattie mitocondriali colpiscono i
mitocondri - piccole fabbriche di
energia che si trovano dentro quasi
tutte le nostre cellule.
I mitocondri sono
responsabili della produzione della
maggior parte di energia necessaria alle
cellule per funzionare. Per questo, essi
sono una fonte così importante di
energia che una tipica cellula umana ne
contiene centinaia. Una malattia
mitocondriale può tagliare fuori da
alcuni a tutti i mitocondri, tagliando
fuori così questa fonte di energia
essenziale.
Quasi tutte le nostre
cellule dipendono dai mitocondri per una
stabile fonte di energia, così una
malattia mitocondriale può trasformarsi
in una malattia multisistemica che
colpisce più di un tipo di cellula,
tessuto o organo. I sintomi non sono
esattamente gli stessi per ognuno,
perché una persona con una malattia
mitocondriale può avere una mistura
unica di mitocondri sani e difettosi,
con una distribuzione unica nel corpo.
Poiché le cellule
muscolari e le cellule nervose hanno un
fabbisogno energetico particolarmente
alto, i problemi muscolari e neurologici
- come la debolezza muscolare,
l'intolleranza all'esercizio, la
sordità, difficoltà di equilibrio e la
coordinazione, attacchi epilettici e
difficoltà di apprendimento -
costituiscono le caratteristiche più
comuni della malattia mitocondriale.
Altre complicazioni frequenti includono
i disturbi della vista, difetti
cardiaci, diabete e stentata crescita.
Solitamente una persona con una malattia
mitocondriale ha due o più di queste
condizioni, alcune delle quali appaiono
associate in forma così regolare da
essere raggruppate all'interno di sindromi.
Una malattia
mitocondriale che causa preminentemente
problemi muscolari è chiamata
miopatia mitocondriale dove (mio
significa muscolo, e pathos
significa malattia), mentre una malattia
mitocondriale che causa sia importanti
problemi muscolari che neurologici è
chiamata encefalomiopatia
mitocondriale (encefalo
si riferisce al cervello).
 |
1. Sistema nervoso
Attacchi epilettici, spasmi,
ritardo nello sviluppo, sordità,
demenza, ictus prima
dei 40 anni, difetti del sistema
visivo, perdita di equilibrio,
problemi con il sistema nervoso
periferico
2.
Occhi Palpebre cadenti
(ptosi), incapacità di muovere gli occhi da parte a parte
(oftalmoplegia esterna), cecità (retinite pigmentosa,
atrofia ottica), cataratte
3.
Cuore
Cardiomiopatia (debolezza del muscolo cardiaco), blocco della
conduzione
4.
Fegato
Insufficienza epatica (poco comune eccetto neonati con
sindrome da deplezione di mtDNA), fegato grasso (steatosi
epatica)
5.
Reni Sindrome di Fanconi
(perdita di metaboliti essenziali nelle urine), sindrome
nefrotica (insolita eccetto che per gli infanti con carenza
di coenzima Q10 ).
6.
Muscoli scheletrici
debolezza muscolare, intolleranza all'esercizio, crampi,
escrezione della proteina muscolare mioglobina nelle urine
(mioglobinuria).
7.
Apparato digestivo
Difficoltà di deglutizione, vomito, sensazione di pienezza,
diarrea cronica, sintomi di ostruzione intestinale
8.
Pancreas Diabete
|
I
principali problemi associati alla
malattia mitocondriale - poca energia,
produzione di radicali liberi ed
acidosi lattica - possono causare
una varietà di sintomi in molti diversi
organi del corpo. Questo diagramma
dipinge i sintomi comuni della malattia
mitocondriale, della quale molte persone
affette manifestano uno specifico
sottoinsieme. Molti di questi sintomi
sono trattabili.
Nonostante ci siano
molti effetti potenziali, le malattie
mitocondriali talvolta causano
disabilità lievi. Talvolta, una persona
ha un sufficiente numero di mitocondri
sani per compensare i difettosi.
Inoltre, poiché molti sintomi della
malattia mitocondriale (come il diabete
o l'aritmia cardiaca) sono comuni nella
popolazione generale, ci sono
trattamenti efficaci per questi sintomi
(tipo l'insulina o i farmaci
anti-aritmia).
Questo opuscolo
descrive le cause generali, le
conseguenze e la gestione delle malattie
mitocondriali, con una enfasi sulle
miopatie e sulle encefalomiopatie ed una
visione dettagliata sulle sindromi più
comuni. Queste includono:
- la sindrome di
Kearns-Sayre (KSS)
- la sindrome di
Leigh
- la sindrome di
deplezione del DNA mitocondriale
(MDS)
-
l'encefalomiopatia mitocondriale,
con acidosi lattica ed episodi di
simil-ictus (MELAS)
- l'epilessia
mioclonica con fibre rosse
sfilacciate (MERRF)
-
l'encefalomiopatia mitocondriale
neurogastrointestinale (MNGIE)
- la neuropatia,
atassia e retinite pigmentosa (NARP)
- la sindrome di
Pearson
- l'oftalmoplegia
esterna progressiva (PEO)
Primo, le malattie
mitocondriali non sono contagiose, e non
sono conseguenti alle azioni di chi le
ha. Esse sono causate da mutazioni, o
alterazioni, nei geni - che pianificano
la produzione delle proteine.
I geni sono
responsabili della costruzione del
nostro corpo, e sono trasmessi dai
genitori ai figli, insieme con tutti i
difetti e le mutazioni che hanno. Questi
significa che le malattie mitocondriali
sono ereditarie, sebbene colpiscano i
membri della stessa famiglia in modi
diversi. (Per maggiori informazioni
sulle mutazioni genetiche e sulla
malattia mitocondriale, vedi "Come
si trasmette nella famiglia?".)
I geni coinvolti nella
malattia mitocondriale normalmente
producono le proteine che lavorano
all'interno dei mitocondri. Entro ogni
mitocondrio (singolare di mitocondri),
queste proteine costituiscono una parte
della linea di assemblaggio che usa
molecole di combustibile derivanti dal
cibo per fabbricare la molecola di
energia ATP. Questo processo di
fabbricazione ad alta efficienza
richiede ossigeno; all'esterno dei
mitocondri, ci sono vie meno efficienti
per produrre ATP senza ossigeno.
Le proteine all'inizio
della linea di assemblaggio
mitocondriale agiscono come portatrici
di carica, importando le molecole
di combustibili - zuccheri e grassi -
all'interno del mitocondrio.
Continuando, altre proteine spezzano gli
zuccheri ed i grassi, estraendo energia
sotto forma di particelle cariche
chiamate elettroni.
 |
|
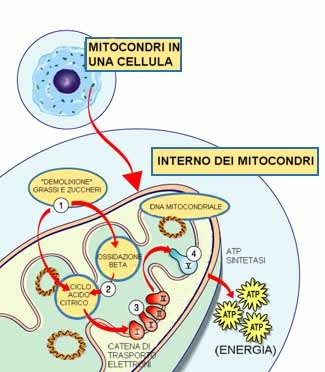
Ogni mitocondrio
è una fabbrica di energia che
“importa” zuccheri e grassi, li
demolisce ossidandoli ed
“esporta” energia (ATP). |
Le proteine verso la
fine della linea - organizzate in cinque
gruppi chiamati complessi I, II, III, IV
e V - utilizzano l'energia di
questi elettroni per produrre ATP. I
complessi dal I al IV trasportano gli
elettroni lungo la l linea e sono perciò
chiamati catena di trasporto degli
elettroni, ed il complesso V chiude
producendo ATP, per questo è chiamato
ATP sintasi.
La causa tipica della
malattia mitocondriale è una carenza in
uno o più di questi complessi
(Infatti, le malattie mitocondriali sono
talvolta chiamate con il nome della
specifica carenza, come nel caso della
carenza del complesso I).
Quando una cellula è
dotata di soli mitocondri difettosi, non
solo si trova ad essere privata di ATP -
in essa vi si possono accumulare un quantitativo
di molecole di combustibile inutilizzate
e ossigeno, con effetti potenzialmente
disastrosi.
In questi casi, le
molecole di combustibile in eccesso
vengono usate per fare ATP in un modo
inefficiente, il quale può generare
sottoprodotti potenzialmente pericolosi
come ad esempio l'acido lattico. (questo
avviene anche quando una cellula ha un
inadeguato apporto di ossigeno, cosa che
può accadere alle cellule muscolari
durante uno esercizio strenuo.)
L'accumulo di acido lattico nel sangue —
chiamato acidosi lattica — è associato
con l'affaticamento muscolare, e può
effettivamente danneggiare il tessuto
muscolare e nervoso.
 |
|
Le miopatie
mitocondriali possono colpire
intere famiglie. |
Contemporaneamente,
l'ossigeno inutilizzato nella cellula
può venire convertito in sostanze
distruttive chiamate specie reattive
dell'ossigeno, comprendenti i cosiddetti
radicali liberi. (Questi sono gli
obiettivi dei cosiddetti farmaci e
vitamine antiossidanti.)
L'ATP prodotta dai
mitocondri costituisce la fonte
principale di energia per la contrazione
delle cellule muscolari e la conduzione
delle cellule nervose. Per questo
motivo, le cellule muscolari e le
cellule nervose sono particolarmente
sensibili ai difetti mitocondriali. Gli
effetti combinati della deprivazione di
energia e dell'accumulazione di tossine
in queste cellule sono probabilmente
all'origine dei principali sintomi delle
miopatie e delle encefalomiopatie
mitocondriali.
I sintomi principali
della miopatia mitocondriale sono
debolezza muscolare, deperimento, e
intolleranza all'esercizio. E'
importante ricordare che la gravità di
ciascuno di questi sintomi varia in
grande misura da una persona ad
un'altra, perfino nella stessa famiglia.
In alcune persone, la
debolezza colpisce in maniera più
pronunciata i muscoli che controllano i
movimenti degli occhi e delle palpebre.
Due conseguenze comuni sono la paralisi
graduale dei movimenti degli occhi,
chiamata oftalmoplegia esterna
progressiva (PEO), e l'abbassamento
delle palpebre superiori, chiamata
ptosi. Spesso, la persona compensa
automaticamente la PEO con movimenti
della testa per guardare nelle diverse
direzioni, e può perfino non rendersi
conto dei problemi alla vista. La ptosi
è potenzialmente più frustrante perché
può ostacolare la visione e causare
anche una mancanza di espressività, ma
può venire corretta chirurgicamente, o
con l'uso di occhiali che abbiano una
"stampella per ptosi" per innalzare le
palpebre superiori.
Le miopatie
mitocondriali possono causare anche
debolezza e deperimento in altri
muscoli della faccia e del collo, che può
portare ad una parlata incomprensibile
ed a difficoltà di deglutizione. In
questi casi, possono essere utili la
logoterapia o dei cambiamenti nella
dieta per facilitare la deglutizione del
cibo. Talvolta, le persone con miopatia
mitocondriale subiscono una perdita
della forza muscolare nelle braccia o
nelle gambe, e possono avere bisogno di
stampelle o di una carrozzina per
muoversi.
L'intolleranza
all'esercizio, detta anche affaticamento
da movimento, si riferisce ad una
inusuale sensazione di esaurimento a
seguito di un esercizio fisico. Il grado
di intolleranza all'esercizio varia
grandemente da persona a persona. Alcune
persone possono avere problemi solamente
con attività atletiche come il jogging,
mentre altre possono avere problemi con
le attività di ogni giorno come camminare
fino alla bussola delle lettere o
sollevare un cartone di latte.
Talvolta, l'intolleranza
all'esercizio è associato con crampi
muscolari dolorosi e/o dolore come
prodotto da ferita. I crampi sono
generalmente una brusca contrazione che
può apparire come un blocco temporaneo
dei muscoli, mentre il dolore come
prodotto da ferita è causato da un
processo acuto di demolizione del
muscolo chiamato
rabdomiolisi, portante a perdita di
mioglobina dei muscoli attraverso le
urine (mioglobinuria). I crampi o la
mioglobinuria sopravvengono generalmente
quando chi ha una intolleranza
all'esercizio “eccede,” e può accadere
durante l'eccessivo esercizio o
parecchie ore più tardi.
 |
|
Alcune persone
con miopatia mitocondriale
perdono la forza nelle loro
gambe e necessitano di una
carrozzina per muoversi. |
Una encefalomiopatia
mitocondriale include tipicamente alcuni
dei sintomi della miopatia
menzionati sopra con in più uno o più
sintomi neurologici. E ancora, questi
sintomi mostrano una vasta gamma di
variabilità individuale sia nel tipo che
nella gravità.
I sintomi più comuni
della encefalomiopatia mitocondriale
sono la perdita di udito, il mal di
testa a grappolo e gli attacchi
epilettici . In almeno una sindrome, il
mal di testa e gli attacchi epilettici
sono spesso accompagnati da episodi di
simil-ictus.
Fortunatamente, ci
sono buoni trattamenti per alcune di
queste condizioni. La perdita di udito
può venire compensata con un apparecchio
acustico e forme di comunicazione
alternative. Spesso, la cefalea può
venire alleviato con farmaci, e gli
attacchi possono essere
prevenuti con dei farmaci usati per
l'epilessia (anti-epilettici).
 |
|
La terapia
occupazionale è importante per i
bambini con una miopatia
mitocondriale. |
Oltre a colpire la
muscolatura degli occhi, una
encefalomiopatia mitocondriale può
colpire gli occhi stessi e le parti del
cervello coinvolte nella visione. Per
esempio, la perdita di visione dovuta
all'atrofia ottica (l'assottigliamento
del nervo ottico) o la retinopatia (la
degenerazione di alcune delle cellule
che si trovano nel fondo dell'occhio)
sono sintomi comuni nella encefalomiopatia mitocondriale. Rispetto
ai problemi muscolari, è più probabile
che questi effetti causino gravi
disturbi della vista.
Spesso,
l'encefalomiopatia mitocondriale causa
atassia, o problemi con l'equilibrio e
con la coordinazione. Le persone con
l'atassia hanno una maggior
predisposizione alle cadute. Si può
ovviare parzialmente a questi problemi
ricorrendo alla terapia fisica ed
occupazionale, e con l'uso di ausili
come dei corrimano, un deambulatore o —
nei casi gravi — una carrozzina.
Talvolta, queste
malattie possono causare una debolezza
importante dei muscoli che supportano la respirazione.
 |
|
La miopatia
mitocondriale può portare a
problemi respiratori che
richiedano l'uso di un
respiratore. |
Altre volte, le
encefalomiopatie mitocondriali causano
anomalie cerebrali che alterano i centri
di controllo del cervello sulla
respirazione.
Una persona con
problemi respiratori minori può
richiedere occasionalmente il ricorso ad
un ausilio respiratorio, come l'aria
pressurizzata, mentre qualcuno con
problemi più gravi può richiedere l'uso
di un respiratore permanente. Coloro che
hanno malattie mitocondriali devono
essere controllati per i segni
dell'insufficienza respiratoria (come il
fiato corto o il mal di testa al
mattino), e lo stato della loro
respirazione deve essere controllato
regolarmente da uno specialista.
Talvolta, le malattie
mitocondriali colpiscono direttamente il
cuore. In questi casi, la causa tipica è
una interruzione del ritmo del battito
cardiaco, chiamato blocco di conduzione.
Sebbene pericolosa, questa condizione è
trattabile con l'uso di un pacemaker, il
quale stimola il normale battito del
cuore. Può venire danneggiato anche il
muscolo cardiaco. Le persone con
malattie mitocondriali possono
necessitare di una esaminazione regolare
da un cardiologo.
 |
|
Nei bambini con
una miopatia mitocondriale può
venire colpito il sistema visivo
del cervello. |
Alcune persone con
malattia mitocondriale hanno seri
problemi renali, problemi
gastrointestinali e/o diabete. Alcuni di
questi problemi sono effetti diretti dei
difetti mitocondriali nei reni, nel
sistema digestivo o nel pancreas
(diabete), e altri sono effetti
indiretti dei difetti mitocondriali in
altri tessuti.
Per esempio, la
rabdomiolisi può portare a problemi
renali a causa di una proteina chiamato
mioglobina prodotta dalla rottura delle
cellule muscolari nella sua escrezione
attraverso le urine. Questa condizione,
chiamata mioglobinuria, costituisce un
sovraccarico per la capacità dei reni a
filtrare le sostanze di rifiuto dal
sangue nelle urine, e può causare danni
renali.
 |
|
Alcuni bambini
con miopatia mitocondriale hanno
ritardi nello sviluppo. |
Visione: Sebbene la
PEO e la ptosi causino tipicamente
solamente un danno visivo leggero negli
adulti, esse sono potenzialmente più
pericolose nei bambini con miopatie
mitocondriali.
Poiché lo sviluppo del
cervello è sensibile alle esperienze nell'età
infantile, la PEO o la ptosi durante
l'età infantile possono talvolta causare
danni permanenti nel sistema visivo del
cervello. Per questa ragione, è
importante che ai bambini con segni
della PEO o della ptosi sia fatto un
controllo specialistico della vista.
Ritardi dello
sviluppo: Dovuto alla debolezza
muscolare, ad anomalie cerebrali o a
combinazione di entrambi, i bambini con
malattie mitocondriali possono avere
difficoltà nello sviluppare certe
capacità. Per esempio, possono impiegare
un tempo insolitamente lungo per
raggiungere pietre miliari motorie come
il sedere, il gattonare e il camminare.
Con il crescere, possono essere incapaci
di andare in giro con la medesima
facilità degli altri bambini loro
coetanei, e possono avere problemi di
linguaggio e/o difficoltà di
apprendimento. I bambini che sono
colpiti gravemente da questi problemi
possono trarre beneficio dai servizi
come la fisioterapia, la logoterapia e
possibilmente da un programma di
istruzione individualizzato (IEP) a
scuola.
|
Le genetiche
mitocondriali sono complesse, e
spesso, si può avere difficoltà
a rintracciare una malattia
mitocondriale nell'albero
genealogico della famiglia. Ma
visto che esse sono causate da
geni difettosi, le malattie
mitocondriali si trasmettono nella
famiglia.
Per capire
come le malattie mitocondriali si
trsmettono nelle famiglie, è
importante sapere che ci sono
due tipi di geni essenziali ai
mitocondri. Il primo tipo è
contenuto dentro il nucleo — la
parte delle nostre cellule che
contiene la maggior parte del
nostro materiale genetico, o
DNA. Il secondo tipo risiede
esclusivamente dentro il DNA
contenuto all'interno dei
mitocondri stessi.
Sia le
mutazioni nel DNA nucleare
(nDNA) che quelle nel DNA
mitocondriale (mtDNA) possono
causare una malattia
mitocondriale.
La maggior
parte del nDNA (insieme con
tutte le mutazioni che ha) viene
ereditato secondo il modello
Mendeliano, intendendo
approssimativamente che
una copia di ogni gene proviene
da un genitore. Inoltre, la
maggior parte delle mutazioni
del nDNA che causano malattie
mitocondriali (comprese la
sindrome di Leigh, la MNGIE e
perfino la MDS) sono autosomiche
recessive, significando che ci
devono essere mutazioni in
entrambe le copie di un gene per
causare la malattia.
Diversamente
dal nDNA, il mtDNA viene
trasmesso al bambino solamente
dalla madre. Questo perché
durante il concepimento, quando
lo sperma si fonde con l'uovo, i
mitocondri dello sperma — ed il
loro mtDNA — vengono distrutti.
Perciò, le malattie
mitocondriali causate da
mutazioni del mtDNA sono
uniche perché sono ereditate
secondo un modello ad
ereditarietà materna (vedi
l'illustrazione sottostante).
Un'altra
caratteristica unica della
trasmissione del mtDNA ha
origine dal fatto che una tipica
cellula umana — compresa la
cellula uovo — contiene
solamente un nucleo, ma
centinaia di mitocondri. Una
singola cellula può contenere
sia mitocondri mutanti che
mitocondri normali, ed è il
rapporto fra i due tipi a
determinare lo stato di salute
della cellula.
Questo aiuta a
spiegare perché i sintomi della
malattia mitocondriale possono
variare così tanto da persona a
persona, perfino all'interno
della stessa famiglia.
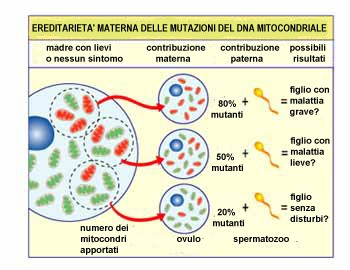
Immaginiamo
che le cellule uovo della donna
(e le altre cellule nel suo
corpo) contengano sia mitocondri
normali che mitocondri mutanti,
e che alcune abbiano solo alcuni
mitocondri mutanti, mentre altre
ne hanno molti. Un bambino
concepito con una cellula uovo
“prevalentemente sana”
probabilmente non svilupperà la
malattia, ed un bambino
concepito con una cellula uovo
“in prevalenza mutante”
probabilmente lo farà.
Inoltre, la
donna potrà essa stessa avere o
non avere i sintomi della
malattia mitocondriale.
Il rischio di
trasmettere una malattia
mitocondriale ai bambini dipende
da molti fattori, compreso se la
malattia è causata da mutazioni
nel nDNA o nel mtDNA.
Una buona
strada per saperne di più
riguardo questo rischio è
parlarne con un dottore o con un
consigliere genetico della
vostra clinica locale della MDA.
Inoltre, vedi vedi il libretto
della MDA “La
realtà delle malattie genetiche
e neuromuscolari.”
 |
|
La
gravità di una malattia
mitocondriale in un
bambino dipende dalla
percentuale dei
mitocondri anormali
(mutanti) nella cellula
uovo da cui ha origine. |
|
Mentre le miopatie
mitocondriali e le encefalomiopatie sono
relativamente rare, alcune delle loro
potenziali manifestazioni sono comuni
nella popolazione generale.
Conseguentemente, per queste
complicazioni (compresi i problemi
cardiaci, gli ictus, gli attacchi
epilettici, le emicranie, la sordità ed
il diabete) ci sono trattamenti
altamente efficaci (comprendenti
farmaci, adattamenti dietetici e
cambiamenti dello stile di vita). (Vedi
“Aspetti
speciali nei bambini.”)
E' una fortuna che
questi sintomi trattabili siano spesso
le maggiori complicazioni da trattare a
vita della malattia mitocondriale.
Tenendo a mente questo, le persone
affette da malattie mitocondriali
possono trarre grandi vantaggi dal controllo della propria
salute e programmando regolari esami
medici.
Invece di focalizzarsi
sulle specifiche complicazioni della
malattia mitocondriale, alcuni
trattamenti mai comprovati mirano a
ovviare o bypassare i mitocondri
difettosi. Questi trattamenti si basano
sull'integrazione dietetica con tre
sostanze naturali coinvolte nella
produzione di ATP nelle nostre cellule.
Sebbene queste non
funzionino per tutti, esse sono di aiuto
per alcuni. (Prenderle sempre sotto
controllo del vostro medico per vedere
cosa sia meglio per la vostra
condizione.)
Una sostanza, la
creatina, normalmente agisce come una
riserva per l'ATP con la formazione di
un composto chiamato fosfato di
creatina. Quando la domanda di ATP di
una cellula eccede la quantità che i
suoi mitocondri possono produrre, la
creatina può rilasciare fosfato (la “P”
in ATP) per migliorare rapidamente la
disponibilità di ATP. Normalmente, il
fosfato di creatina (chiamato anche
fosfocreatina) fornisce tipicamente
l'impulso iniziale di ATP necessario per
l'attività muscolare intensa.
Un'altra sostanza, la
carnitina, generalmente migliora
l'efficienza della produzione di ATP
aiutando l'importazione di alcuni tipi
di molecole di combustibile dentro i
mitocondri, e rimuovendo alcuni dei
sottoprodotti tossici della produzione
di ATP. La carnitina è disponibile come
integratore su ricetta chiamato
L-carnitina.
 |
|
Le miopatie
mitocondriali possono venire
ereditate, e la gravità può
variare dentro una famiglia. |
Infine, il coenzima Q10,
o coQ10, è un componente
della catena di trasporto degli
elettroni, la quale usa l'ossigeno per
produrre ATP. Alcune malattie
mitocondriali sono causate dalla carenza
di coQ10, e ci sono buone
evidenze che l'integrazione di coQ10
è di beneficio in questi casi. Alcuni
medici pensano che l'integrazione di
coQ10 possa alleviare anche altre
malattie mitocondriali.
L'integrazione di
creatina, L-carnitina e coQ10
sono spesso combinate in un “cocktail”
per il trattamento delle malattie
mitocondriali. Sebbene ci siano poche
prove scientifiche che questo
trattamento funzioni, molte persone con
malattia mitocondriale hanno riferito di
modesti benefici. Prima di assumere
qualsiasi farmaco o integratore dovreste
consultare il vostro dottore o il
direttore della clinica della MDA.
Nota: Tipicamente,
queste sindromi sono ereditate o in un
modello materno o in un cosiddetto
modello mendeliano, e/o esse sono
sporadiche, che significa manifestate
senza una storia familiare. Per maggiori
informazioni riguardo l'ereditarietà,
vedi “Come si
trasmettono nella famiglia?”.
Modello
dell'ereditarietà:
sporadica
Insorgenza: prima dei 20 anni
Caratteristiche: Questo
disturbo è definito come PEO
(solitamente ne è il sintomo iniziale) e
retinopatia pigmentosa, una
pigmentazione a “sale e pepe” nella
retina che può colpire la visione, ma
che spesso la lascia intatta.
Altri sintomi comuni
includono il blocco di conduzione (nel
cuore) e l'atassia. Sintomi meno tipici
sono il ritardo o il deterioramento
mentale, il ritardo nella maturazione
sessuale e la bassa statura.
Modello
dell'ereditarietà:
materna, Mendeliana
Insorgenza: infanzia
Caratteristiche: la sindrome di
Leigh causa anomalie cerebrali che
possono provocare atassia, attacchi
epilettici, danneggiamento della visione
e dell'udito, ritardo mentale ed un
controllo alterato sulla respirazione.
Essa causa anche
debolezza muscolare, con effetti
preminenti sulla deglutizione, la parola
ed i movimenti oculari.
Modello
dell'ereditarietà:
Mendeliana
Insorgenza: infanzia
Caratteristiche: Questo
disturbo tipicamente causa debolezza
muscolare e/o insufficienza epatica, e
più raramente, anomalie cerebrali.
“Rammollimento,” difficoltà di
alimentazione, e ritardo mentale sono
sintomi comuni; la PEO e gli attacchi
epilettici sono meno comuni.
Modello
dell'ereditarietà:
materna
Insorgenza: dall'età infantile
all'inizio dell'età adulta
Caratteristiche: la MELAS causa
ricorrenti episodi di simil-ictus nel
cervello, il mal di testa a grappolo,
vomito e attacchi epilettici, e può
portare a danni cerebrali permanenti.
Altri sintomi comuni includono la PEO,
debolezza muscolare generalizzata,
intolleranza all'esercizio, perdita di
udito, diabete e bassa statura.
Modello
dell'ereditarietà:
materna
Insorgenza: dalla tarda età
infantile all'adolescenza
Caratteristiche: I sintomi
preminenti sono i miocloni (spasmi muscolari), gli attacchi epilettici,
l'atassia e la debolezza muscolare. La
malattia può anche causare perdita di
udito e bassa statura.
Modello
dell'ereditarietà:
Mendeliana
Insorgenza: solitamente prima
dei 20 anni
Caratteristiche: Questo
disturbo causa PEO, ptosi, debolezza
degli arti e problemi gastrointestinali
(digestivi), comprendenti la diarrea
cronica ed il dolore addominale. Un
altro sintomo comune è la neuropatia
periferica (un malfunzionamento dei
nervi che può portare a danni sensori e
debolezza muscolare).
Modello
dell'ereditarietà:
materna
Insorgenza: dall'infanzia
all'età adulta
Caratteristiche: la NARP causa
neuropatia (vedi sopra), atassia e
retinite pigmentosa (una degenerazione
della retina degli occhi, che provoca
una perdita della visione). Può causare
anche ritardo nello sviluppo, attacchi
epilettici e demenza.
Modello
dell'ereditarietà:
sporadica
Insorgenza: infanzia
Caratteristiche: Questa
sindrome causa grave anemia e
malfunzionamento del pancreas. I bambini
che sopravvivono a questa malattia
solitamente sviluppano la KSS.
Modello
dell'ereditarietà:
materna, Mendeliana, sporadica
Insorgenza: Solitamente
nell'adolescenza o nella prima età
adulta
Caratteristiche: Come riportato
sopra, la PEO è spesso un sintomo della
malattia mitocondriale, ma talvolta si
presenza come una sindrome distinta.
Spesso, è associata con l'intolleranza
all'esercizio.
Nessuno dei sintomi
che caratterizzano le malattie
mitocondriali — debolezza muscolare,
intolleranza all'esercizio, perdita di
udito, atassia, attacchi epilettici,
difficoltà di apprendimento, cataratte,
difetti cardiaci, diabete e crescita
stentata — sono unici delle malattie
mitocondriali. Comunque, una persona che
presenti una combinazione di tre o più
di questi sintomi costituisce una forte
indicazione di malattia mitocondriale,
specialmente quando i sintomi
coinvolgono più di un sistema di organi.
Per valutare
l'estensione di questi sintomi, un
medico solitamente inizia con il sentire
la storia medica personale del paziente,
e prosegue quindi con gli esami
fisiologici e neurologici.
L'esame fisiologico
include tipicamente le prove di forza e
di resistenza, ed una prova di
esercizio, la quale può comprendere
attività come aprire e chiudere
ripetitivamente il pugno, o il salire e
scendere una piccola scala. L'esame
neurologico può includere gli esami dei
riflessi, della visione, della
verbalizzazione e delle abilità
cognitive di base (pensiero).
A seconda delle
informazioni reperite durante l'anamnesi
(la storia personale) e dagli esami, il
medico può procedere con esami più
specialistici che possono individuare
anormalità nei muscoli, nel cervello e
in altri organi.
Il più importante di
questi esami è la biopsia muscolare, che
consiste nel rimuovere un piccolo
campione di tessuto muscolare per
esaminarlo. Quando viene trattato con un
colorante che colora di rosso i
mitocondri, muscoli affetti da malattia
mitocondriale spesso mostrano fibre
rosse sfilacciate — cellule muscolari
(fibre) che hanno un eccesso di
mitocondri. Altre colorazioni possono
rivelare l'assenza di enzimi
mitocondriali essenziali nei muscoli. E'
possibile anche estrarre proteine
mitocondriali dal muscolo e misurare la
loro attività.
Oltre alla biopsia
muscolare, si possono usare tecniche non
invasive per esaminare il muscolo senza
prelevare campioni di tessuto. Per
esempio, una tecnica chiamata risonanza
magnetica spettroscopica del fosforo
muscolare (MRS) può misurare i livelli
della fosfocreatina e dell'ATP (che sono
spesso carenti nei muscoli affetti da
malattia mitocondriale).
La TC a scansione
(TAC) e la MRI a scansione possono
venire usate per ispezionare visivamente
il cervello per individuare segni di
danni, e la superficie degli elettrodi
sistemati sul cuoio capelluto possono
essere usate per produrre una
registrazione dell'attività del cervello
chiamata elettroencefalogramma (EEG).
Si possono usare
tecniche simili per esaminare le
funzioni di altri organi e tessuti nel
corpo. Per esempio, un
elettrocardiogramma (ECG) può monitorare
l'attività cardiaca, e un esame del
sangue può identificare segni di
malfunzionamento dei reni.
Infine, un esame
genetico può accertare se una persona ha
una mutazione genetica che causa una
malattia mitocondriale. Idealmente,
l'esame è fatto usando materiale
genetico estratto dal sangue o da una
biopsia muscolare. E' importante tenere
presente che, sebbene un risultato
positivo può confermare una diagnosi, un
risultato negativo non è necessariamente
significativo.
|
ESAMI DIAGNOSTICI NELLE MALATTIE MITOCONDRIALI
|
| Tipo |
Esame |
Cosa mostra |
 |
Esami clinici o
storia orale dei membri della famiglia |
Può
talvolta indicare un
modello di ereditarietà
notando per esempio
"segni leggeri" in
parenti non affetti.
Questi includono
sordità, bassa statura,
mal di testa a grappolo
e PEO. |
 |
1. Istochimica
2. Immuno istochimica
3. Biochimica
4. Microscopio elettronico
|
1. Scopre la
proliferazione anormale di mitocondri e la carenza di
citocromo C ossidasi (COX, che è nel complesso IV della catena
di trasporto degli elettroni).
2. Scopre la presenza o l'assenza di specifiche proteine.
Può escludere altre malattie o confermare la perdita di
proteine nella catena di trasporto degli elettroni.
3. Misura l'attività di specifici enzimi. Un esame speciale
chiamato polarografia misura il consumo di ossigeno nei
mitocondri.
4. Può confermare l'aspetto anormale dei mitocondri. Non si
usa più molto al giorno d'oggi.
|
 |
1. Livelli del
lattato e del piruvato
2. Creatina chinasi serica
|
1. Se elevati,
possono indicare difetti nella catena di trasporto degli
elettroni; il rapporto anormale fra i due può aiutare a
identificare la parte della catena che è bloccata.
2. Può essere leggermente elevata nelle malattie
mitocondriali ma solitamente solo in caso di deplezioni del
DNA mitocondriale elevate.
|
 |
1. Mutazioni
conosciute
2. Mutazioni rare o sconosciute
|
1. Usa campioni di
sangue o di muscolo per cercare mutazioni conosciute, guardando
prima alle mutazioni comuni.
2. Può inoltre cercare mutazioni rare o sconosciute ma
può richiede campioni dai membri della famiglia; questa è molto più
costosa e richiede tempi lunghi.
|
|
Con l'appoggio della MDA, gli
scienziati continuano a fare progressi
significativi nella loro ricerca di
comprendere completamente e trattare le
malattie mitocondriali.
Un progresso unico è stato fatto
nella
MNGIE, una malattia nella quale un
difetto in un gene nel nucleo causa
indirettamente un problema
mitocondriale. I ricercatori
finanziati dalla MDA stanno
sperimentando con l'infusione di cellule
staminali da donatore nei pazienti con
la MNGIE di recuperare le normali
condizioni metaboliche e fermare il
danneggiamento dei mitocondri.
In aggiunta, gli scienziati
finanziati dalla MDA hanno identificato
molti dei difetti genetici che causano
le malattie mitocondriali. Essi hanno
usato le conoscenze di quei difetti
genetici per creare dei modelli animali
della malattia mitocondriale, i quali
possono essere usati per studiare dei
potenziali trattamenti. Essi hanno anche
ideato degli esami genetici che
permettono accurate diagnosi dei difetti
mitocondriali e forniscono informazioni
affidabili per la pianificazione
famigliare.
Forse più importante, è che la
conoscenza dei difetti genetici che
causano la malattia mitocondriale apre
alla possibilità che un giorno vengano
sviluppati trattamenti che mirano a
questi.
| Il sito web
della MDA viene aggiornato
costantemente con le ultimissime
informazioni riguardo le
malattie nel suo programma. Vedi
le ultimissime
novità dalla ricerca. |
Recentemente, gli scienziati hanno
scoperto che un peptide chiamato TAT,
quando è attaccato ad una proteina
terapeutica o ad un gene, può portare il
suo carico dentro i mitocondri e
mantenervelo.
Dal 2008, gli
scienziati supportati dalla MDA hanno
allevato topi con malattie
mitocondriali e stanno continuando a
sviluppare nuovi tipi di queste topi, come
pure pesci e uccelli con difetti
genetici colpenti i mitocondri.
Alcuni ricercatori finanziati dalla MDA stanno studiando
come il DNA mitocondriale viene convertito
(trascritto) in RNA e come
l'RNA mitocondriali viene convertito
(traslato) nelle proteine
mitocondriali, con il fine ultimo di
correggere queste processi quando esse
sono difettosi.
Altri stanno studiando i percorsi
biochimici che permettono ai mitocondri
di regolare i livelli del rame; altri come una
proteina che aumenta l'attività dei
geni correlati alla funzione mitocondriale
potrebbe avere applicazione nel
trattamento della malattia; altri come un gene nel cromosoma
11 potrebbe regolare la proliferazione
dei
mitocondri all'interno delle cellule
muscolari; e altri ancora cosa avviene quando
nei mitocondri una molecola sensibile
all'ossigeno non lavora.
La Muscular Dystrophy Association
offre a vasta gamma di
servizi per aiutare te e la tua
famiglia ad affrontare la
miopatia mitocondriale. Se sei un
adulto che ha appena ricevuto una
diagnosi, o sei il genitore di un
bambino con a miopatia mitocondriale, lo
staff del tuo
ufficio locale della MDA è qui per
assisterti in molti modi. I servizi
dell'associazione comprendono:
- una rete di ampiezza nazionale
di
220
cliniche affiliate ad ospedali
provviste dei migliori specialisti
nelle malattie neuromuscolari
- I
campi estivi della MDA per
bambini con malattie neuromuscolari
- gruppi di supporto
professionalmente facilitati per chi
è affetto, i coniugi, i genitori o
altri prestatori di cure
- assistenza l'acquisto e la
riparare di carrozzine, ausili per
gli arti, e dispositivi di
comunicazione
- Esami valutativi per terapie
fisiche, occupazionali, logopedistiche
e respiratorie
- vaccinazioni antiinfluenzali per
aiutare a proteggere il sistema
respiratorio
- noleggio di ausili ed
apparecchiature
Il programma pubblico di educazione
alla salute della MDA ti aiuterà a
mantenere il passo con le novità dalla
ricerca, le scoperte mediche e le
informazioni sulla disabilità attraverso
relatori didattici, seminari, video,
bollettini ed altro. Abbi fiducia e
chiedi al tuo ufficio locale gli ultimi
opuscoli della MDA, compreso il “Servizi
per la persona, la famiglia e la
comunità.” Molti opuscoli della MDA
sono disponibili in spagnolo.
La rivista
Quest, della MDA premiata con il
premio award. Quest pubblica articoli
dettagliati riguardo le scoperte della
ricerca; le cure mediche e quotidiane; i
prodotti ed i dispositivi utili; i
problemi sociali e famigliari; e molto
di più.
Se hai delle domande riguardo le
miopatie mitocondriali, qualcuno alla
MDA ti aiuterà a trovare la risposta.
La realtà
delle miopatie mitocondriali
|

Ritorna al
fascicolo delle
malattie
|









