|
Malattia |
Caratteristiche cliniche principali |
Difetto molecolare |
Ereditarietà |
Esami diagnostici consigliati |
|
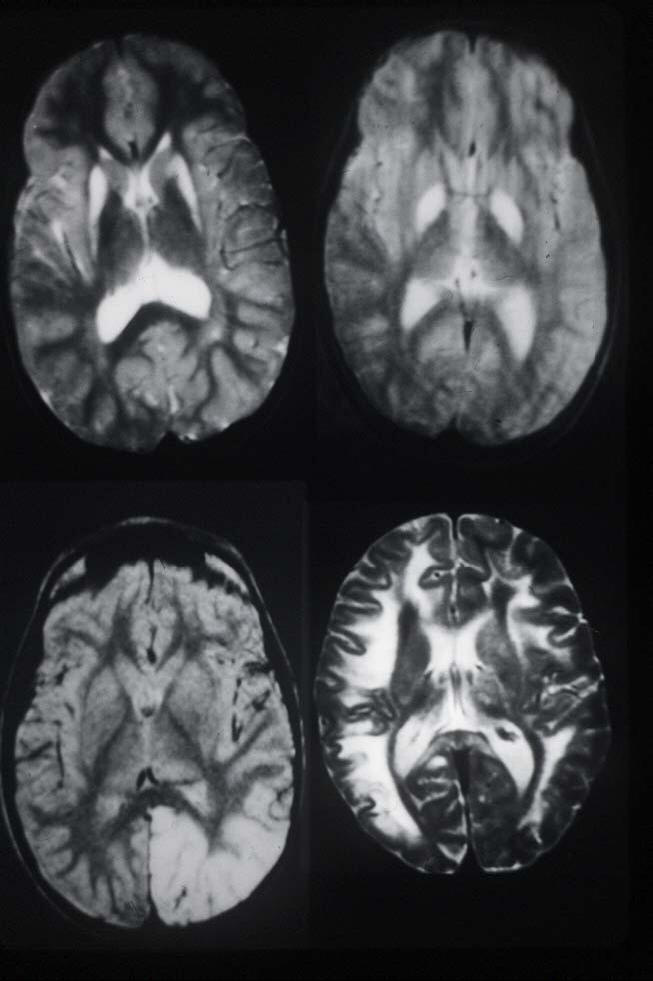
Sindrome di Leigh |
Regressione psicomotoria +/-ptosi, oftalmoparesi,
retinopatia, attacchi epilettici, atassia, neuropatia periferica,
cardiomiopatia, insufficienza epatica |
Nucleare o mtDNA |
M o AR |
MRI del cervello; acido lattico nel sangue, CSF, o entrambi;
biopsia muscolare; screening focalizzato sulle mutazioni sulla
base dei difetti negli enzimi della catena respiratoria |
|
Sindrome di Kearns Sayre (KSS) |
Debolezza
muscolare extraoculare, retinopatia pigmentosa ,
e
insorgenza prima dell'età di 20 anni, più almeno una delle seguenti:
blocco di conduzione cardiaca, atassia, CSF proteine >100 mg/dl |
delezione nel mtDNA |
S |
ECG; acido lattico nel sangue, CSF, o entrambi;
esaminazione oftalmologica ; audiogramma;
biopsia muscolare |
|
Oftalmoplegia esterna progressiva
cronica (CPEO) |
Debolezza extraoculare muscolare (ptosi, oftalmoparesi), +/-
disfagia, debolezza prossimale degli arti |
delezione nel mtDNA o mutazione puntiforme. |
S o M |
ECG; acido lattico nel sangue, CSF, o entrambi;
esaminazione oftalmologica ; audiogramma; se
sporadica, biopsia muscolare; se ereditata maternamente, screening della
mutazione del mtDNA nel sangue |
|
Encefalomiopatia mitocondriale
acidosi lattica e episodi di simil-ictus (MELAS) |
Episodi di simil-ictus, attacchi epilettici, demenza, +/- perdita di udito,
retinopatia, diabete mellito, cardiomiopatia, dismotilità gastrointestinale |
mtDNA 3243A>G o
altre mutazioni nel mtDNA |
M |
MRI del cervello; acido lattico nel sangue; audiogramma; se
ereditata maternamente , screening della mutazione del mtDNA
nel sangue; esaminazione cardiaca |
|
Epilessia mioclonica, fibre rosse sfilacciate (MERRF) |
Miocloni, epilessia, atassia, miopatia, +/- encefalopatia,
lipomi, neuropatia periferica, atrofia ottica, diabete |
mtDNA 8344A>G o
altri mutazioni nel mtDNA |
M |
MRI del cervello; acido lattico
nel sangue; audiometria; se
ereditata maternamente, screening della mutazione del mtDNA nel sangue |
|
Neuropatia, atassia, retinite pigmentosa (NARP) |
Neuropatia periferica, atassia, retinite pigmentosa |
mtDNA 8993T>G o
altre mutazioni nel mtDNA |
M |
MRI del cervello; acido lattico
nel sangue; EMG/NCS;
esaminazione oftalmologica; screening della mutazione del mtDNA nel sangue |
|
Neuropatia ottica ereditaria di Leber
(LHON) |
Perdita di visione
subacuta solitamente unilaterale seguita da perdita di visione
nel secondo occhio settimane o mesi dopo. Teleangectasia peripapillaria. Ereditarietà materna, principalmente colpente uomini giovani |
mtDNA 11778G>A,
3460G>A, 14484T>C |
M |
Esaminazione oftalmologica; ECG; screening
della mutazione del mtDNA nel sangue |
|
Sordità associata a aminoglicosidi o perdita di udito non-sindromica |
Perdita di udito neurosensoria,
ereditarietà materna |
mtDNA 1555A>G |
M |
Audiogramma e altri esami auditivi;
screening della mutazione del mtDNA nel sangue |
|
PEO autosomica dominante con
delezioni multiple del mtDNA |
Ptosi, oftalmoparesi |
POLG1, PEO1, ANT1,
POLG2 |
AD |
Esaminazione oftalmologica;
sequenziazione dei geni POLG1, PEO1,
ANT1 e POLG2 |
|
PEO autosomica recessiva con
delezioni multiple del mtDNA |
Ptosi, oftalmoparesi |
POLG1 |
AR |
Esaminazione oftalmologica;
sequenziazione del gene POLG1
|
|
Encefalopatia
neurogastrointestinale mitocondriale
(MNGIE) |
Ptosi, oftalmoparesi, neuropatia periferica,
dismotilità
gastrointestinale, cachessia, leucoencefalopatia,
+/- perdita di udito |
TYMP |
AR |
Esaminazioni
oftalmologica e gastrointestinale; NCS/EMG; MRI del cervello; audiogramma;
timidina
plasmatica e desossiuridina; attività della timidina fosforilasi buffy coat;
sequenziazione del gene TYMP
|
|
Deplezione del mtDNA, forma miopatica |
Miopatia ad insorgenza Infantile o nella fanciullezza, acidosi lattica |
TK2 |
AR |
NCS/EMG; acido lattico
nel sangue; CK serico; biopsia muscolare; EEG;
sequenziazione del gene TK2 |
|
Deplezione del mtDNA, forma epatica o
epatocerebrale (compresa la
sindrome di Alpers) |
Epatopatia ad insorgenza infantile o nella fanciullezza e encefalopatia,
acidosi lattica |
DGUOK, SUCLA2,
MPV17, POLG1,
SUCLG1 |
AR |
Acido lattico
nel sangue; panel enzimi epatici; EEG;
DGUOK, SUCLA2, SULG1, sequenziazione del gene POLG1 |
|
Atrofia
ottica autosomica dominante
o Atrofia ottica autosomica dominante con perdita di udito, e CPEO |
Atrofia ottica lentamente progressiva +/- perdita di udito neurosensoria , ptosi, e oftalmoparesi |
OPA1 |
AD |
Esaminazione oftalmologica; audiogramma, sequenziazione del gene OPA1
|
|
sindrome Mohr-Tranebjaerg |
Distonia, cecità corticale, delusioni paranoidee. |
TIMM8A |
XLR |
Esaminazione oftalmologica; audiogramma; sequenziazione del gene
TIMM8A
|