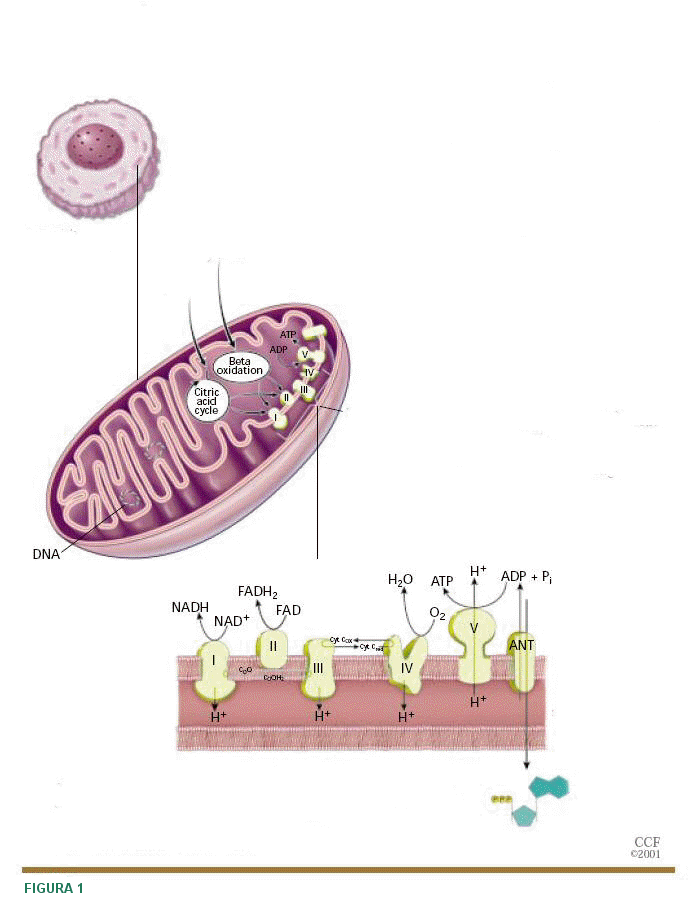
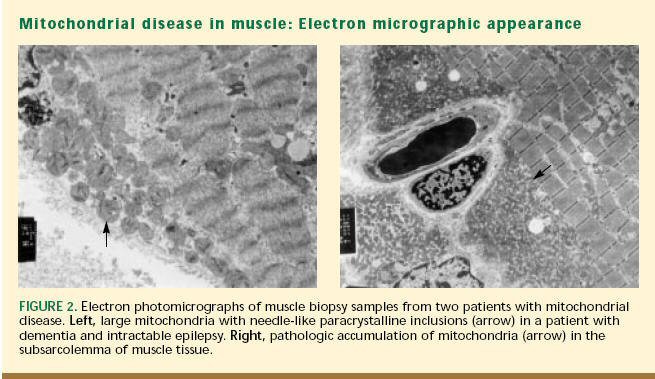
|
██ RIFERIMENTI
-
Luft R, Ikkos D,
Palmieri G, Ernster L, Afzelius B. A case of severe
hypermetabolism of nonthyroid origin with a defect in the
maintenance of mitochondrial respiratory control: A
correlated clinical, biochemical and morphological study. J
clin Invest 1962; 41:1776–1804.
-
DiMauro S,
Bonilla E. Mitochondrial encephalomyopathies. In: Rosenberg
RN, Prusiner SB, DiMauro S, Barchi RL, editors. The
molecular and genetic basis of neurological disease. Boston:
Butterworth-Heinemann, 1997:201–235.
-
Shoffner JM,
Wallace Dc. Oxidative phosphorylation diseases. In: Scriver
cR, Beaudet AL, Sly WS, et al, editors. The metabolic and
molecular bases of inherited disease. New York: McGraw-Hill,
1995:1535–1610.
-
Wallace Dc.
Mitochondrial diseases in man and mouse. Science 1999;
238:1482–1488.
-
Giles RE, Blanc H, cann HM, Wallace Dc. Maternal inheritance
of human mitochondrial DNA. Proc Natl Acad Sci USA 1980;
77:6715–6719.
-
Shoffner JM.
Mitochondrial myopathy diagnosis. Neurol clin 2000;
18:105–123.
-
Andreu AL, Hanna MG, Reichmann H, et al. Exercise
intolerance due to mutations in the cytochrome b gene of
mitochondrial DNA. N Engl J Med 1999; 341:1037–1044.
-
Fischel-Ghodsian N, Prezant TR, chaltraw WE, et al.
Mitochondrial gene mutation is a significant predisposing
factor in aminoglycoside ototoxicity. Am J Otolaryngol 1997;
18:173–178.
-
corral-Debrinski
M, Shoffner JM, Lott MT, Wallace Dc. Association of
mitochondrial DNA damage with aging and coronary
atherosclerotic heart disease. Mutat Res 1992; 275:169–180.
-
Wallace Dc. Mitochondrial DNA mutations and bioenergetic
defects in aging and degenerative diseases. In: Rosenberg
RN, Prusiner SB, DiMauro S, Barchi RL, editors. The
molecular and genetic basis of neurologic disease. Boston,
Butterworth-Heinemann, 1997:237–269.
-
Melov S, Shoffner JM, Kaufman A, Wallace Dc. Marked increase
in the number and variety of mitochondrial DNA
rearrangements in aging human skeletal muscle. Nucleic Acids
Res 1995; 23:4122–4126.
- Pavlakis SG, Phillips Pc, DiMauro S, DeVivo Dc, Rowland
LP. Mitochondrial myopathy, encephalomyopathy, acidosi lattica and strokelike episodes: a distinctive clinical
syndrome. Ann Neurol 1984; 16:481–488.
- Goto Y, Nonaka I, Horai S. A mutation in the tRNALeu(URR)
gene associated with the MELAS subgroup of mitochondrial
encephalomyopathies. Nature 1990; 348:653.
-
Bardosi A, creutzfeldt W, DiMauro S, et al. Myo-, neuro-,
gastrointestinal encephalomyopathy (MNGIE syndrome) due to
partial deficiency of cytochrome c oxidase: a new
mitochondrial multisystem disorder. Acta Neuropathol (Berl)
1987; 74:248–158.
-
Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase
gene mutations in MNGIE, a human mitochondrial disorder.
Science 1999; 283:689–692.
- Lietman PS. Mitochondrial protein synthesis: inhibition
by emetine hydrochloride. Mol Pharmacol 1971; 7:122–128.
- Huang cc, chu cc, Pang cY, Wei YH. Tissue mosaicism in
the skeletal muscle and sural nerve biopsies in the MELAS
syndrome. Acta Neurol Scand 1999; 99:125–129.
- Peyronnard JM,
charron L, Bellavance
A, Marchand L. Neuropathy
and mitochondrial myopathy. Ann Neurol 1980; 7:262–268.
- Kenny D, Wetherbee J.
Kearns-Sayre syndrome in the elderly: mitochondrial myopathy
with advanced heart block. Am Heart J 1990; 120:440–443.
- Mashima Y,
Kigasawa K, Hasegawa H, Tani M, Oguchi Y. High incidence of
pre-excitation syndrome in Japanese families with Leber’s
hereditary optic neuropathy. clin Genet 1996; 50:535–537.
- Kerrison JB,
Biousse V, Newman NJ. Retinopathy of NARP syndrome. Arch
Ophthalmol 2000; 118:298–299.
- Smith PR,
Bain Sc, Good PA, et al. Pigmentary retinal dystrophy and
the syndrome of maternally inherited diabetes and deafness
cause by the mitochondrial DNA 3243 tRNA (Leu) A to G
mutation. Ophthalmology 1999; 106:1101–1108.
- Wallace Dc,
Singh G, Lott MT, et al. Mitochondrial DNA mutation
associated with Leber’s hereditary optic neuropathy. Science
1988; 242:1427–1430.
- chalmers RM,
Schapira AH. clinical, biochemical and molecular genetic
features of Leber’s hereditary optic neuropathy. Biochim
Biophys Acta 1999; 1410:147–158.
-
Fischel-Ghodsian N. Mitochondrial genetics and hearing loss:
the missing link between genotype and phenotype. Proc Soc
Exp Biol Med 1998; 218:1–6.
- Jacobs HT.
Mitochondrial deafness. Ann Med 1997; 29:483–491.
- Niaudet P.
Mitochondrial disorders and the kidney. Arch Dis child 1998;
78:387–390.
- Niaudet P,
Rotig A. The kidney in mitochondrial cytopathies. Kidney Int
1997; 51:1000–1007.
- Odawara M.
Involvement of mitochondrial gene abnormalities in the
pathogenesis of diabetes mellitus. Ann NY Acad Sci 1996;
786:72–81.
- Gerbitz KD,
van den Ouweland JM, Maassen JA, Jaksch M. Mitochondrial
diabetes mellitus: a review. Biochim Biophys Acta 1995;
1271:253–260.
- van den
Ouweland JM, Lemkes HH, Trembath Rc, et al. Maternally
inherited diabetes and deafness is a distinct subtype of
diabetes and associates with a single point mutation in the
mitochondrial tRNA (Leu(URR)) gene. Diabetes 1994;
43:746–751.
- Plioplys AV,
Plioplys S. Serum levels of carnitine in chronic fatigue
syndrome: clinical correlates. Neuropsychobiology 1995;
32:132–138.
- Kuratsune H,
Yamaguti K, Takahashi M, Tagawa S, Kitani T. Acylcarnitine
deficiency in chronic fatigue syndrome. clin Infect Dis
1994; 18(suppl 1):62–67
- Griggs Rc,
Karpati G. Muscle pain, fatigue, and mitochondriopathies. N
Engl J Med 1999; 341:1077–1078.
- Walker UA,
collins S, Byrne E. Respiratory chain encephalomyopathies: a
diagnostic classification. Eur Neurol 1996; 36:260–267.
- Goda S,
Hamada T, Ishimoto S, Kobayashi T, Goto I, Kuroiwa Y.
clinical improvement after administration of coenzyme Q10 in
a patient with mitochondrial encephalomyopathy. J Neurol
1987; 234:62–63.
- Ogasahara S,
Yorifuji S, Nishikawa Y, et al. Improvement of abnormal
pyruvate metabolism and cardiac conduction defect with
coenzyme Q10 in Kearns-Sayre syndrome. Neurology 1985;
35:372–377.
- Bresolin N,
Bet L, Binda A, et al. clinical and biochemical correlations
in mitochondrial myopathies treated with coenzyme Q10.
Neurology 1988; 38:892–899.
- Abe K,
Fujimura H, Nishikawa Y, et al. Marked reduction in cSF
lactate and pyruvate levels after coQ therapy in a patient
with mitochondrial myopathy, encephalopathy, acidosi lattica
and stroke-like episodes (MELAS). Acta Neurol Scand 1991;
83:356–359.
- Gold R,
Seibel P, Reinelt G, et al. Phosphorous magnetic resonance
spectroscopy in the evaluation of mitochondrial myopathies:
Results of a 6month therapy study with coenzyme Q. Eur
Neurol 1996; 36:191–196.
- Barbiroli B,
Iotti S, Lodi R. Improved brain and muscle mitochondrial
respiration with coQ. An in vivo study by 31P-MR
spectroscopy in patients with mitochondrial cytopathies.
BioFactors 1999; 9:253–260.
- Bendahan D,
Desnuelle c, Vanuxem D, et al. 31P NMR spectroscopy and
ergometer exercise test as evidence for muscle oxidative
performance improvement with coenzyme Q in mitochondrial
myopathies. Neurology 1992; 42:1203–1209.
- Bresolin N,
Doriguzzi c, Ponzetto c, et al. Ubidecarenone in the
treatment of mitochondrial myopathies: a multi-center
double-blind trial. J Neurol Sci 1990; 100:70–78.
- Zierz S, von
Wersebe O, Bleistein J, Jerusalem F. Exogenous coenzyme Q (coQ)
fails to increase coQ in skeletal muscle of two patients
with mitochondrial myopathies. J Neurol Sci 1990;
95:283–290.
- Matthews PM,
Ford B, Dandurand RJ, et al. coenzyme Q10 with multiple
vitamins is generally ineffective in treatment of
mitochondrial disease. Neurology 1993; 43:884–890.
- Pons R,
DeVivo Dc. Primary and secondary carnitine deficiency
syndrome. J child Neurol 1995; 10(suppl 2):8–24.
- campos Y,
Huertas R, Lorenzo G, et al. Plasma carnitine insufficiency
and effectiveness of L-carnitine therapy in patients with
mitochondrial myopathy. Muscle Nerve 1993; 16:150–153.
- Harris Rc,
Soderlund K, Hultman E. Elevation of creatine in resting and
exercised muscle of normal subjects by creatine
supplementation. clin Sci 1992; 83:367–374.
- Tarnopolsky
MA, Roy BD, MacDonald JR. A randomized, controlled trial of
creatine monohydrate in patients with mitochondrial
cytopathies. Muscle Nerve 1997; 20:1502–1509.
-
Tarnopolsky MA,
Martin J. creatine monohydrate increases strength in
patients with neuromuscular disease. Neurology 1999;
52:854–857.
-
Naito E, Ito M,
Yokota I, et al. concomitant administration of sodium
dichloroacetate and thiamine in West syndrome caused by
thiamineresponsive pyruvate dehydrogenase complex deficiency.
J Neurol Sci 1999; 171:56–59.
-
DiRocco M, Lamba
LD, Minniti G, caruso U, Naito E. Outcome of thiamine
treatment in a child with Leigh disease due to
thiamine-responsive pyruvate dehydrogenase deficiency. Eur J
Paediatr Neurol 2000; 4:115–117.
-
Gregersen N.
Riboflavin-responsive defects of beta-oxidation. J Inherit
Metab Dis 1985; 8(suppl 1):65–69.
-
Wolf B, Heard
GS, Jefferson LG, Proud VK, Nance WE, Weissbecker KA.
clinical findings in four children with biotinidase
deficiency detected through a statewide neonatal screening
program. N Engl J Med 1985; 313:16–19.
-
Schoenen J,
Jacquy J, Lenaerts M. Effectiveness of high-dose riboflavin
in migraine prophylaxis. A randomized controlled trial.
Neurology 1998; 50:466–470.
-
Yee AJ.
Effectiveness of high-dose riboflavin in migraine
prophylaxis. Neurology 1999; 52:431–432.
-
Mattimoe D,
Newton W. High-dose riboflavin for migraine prophylaxis. J
Fam Pract 1998; 47:11.
-
Sastre J,
Pallardo FV, Garcia de la Asuncion J, Vina J. Mitochondria,
oxidative stress and aging. Free Radic Res 2000; 32:189–198.
-
Mecocci P,
MacGarvey U, Kaufman AE, et al. Oxidative damage to
mitochondrial DNA shows marked age-dependent increases in
human brain. Ann Neurol 1993; 34:609–616.
-
Beal MF. Does
impairment of energy metabolism result in excitotoxic
neuronal death in neurodegenerative illness? Ann Neurol
1992; 31:119–130.
ADDRESS: Bruce H. cohen, MD, chief, Section of Pediatric
Neurology, S80, The cleveland clinic Foundation, 9500 Euclid
Avenue, cleveland, OH 44195.
|