

|
|
Think
Mitocondri


UNITED MITOCHONDRIAL DISEASE FOUNDATION

| Afew anni ago, I had l'opportunità to speak to un gruppo of genitori e dottori circa l'obiettivo del UMDF. Afterwards, uno dei dottori approached me e wanted sapere what medica sfondo I had qualifying me to discuss the approcci necessarie per raggiungere a cure. He realmente got my attention quando he said, “You volunteers sono tutti alike.” I detto him I consider myself a donkey non a volontari. A donkey harnessed to a grande wagon. I detto him non to spend ogni time thinking circa the donkey, ma tell the donkey quale è necessaria to trovano a cure per malattie mitocondriali. I detto him I couldn’t sviluppare the complesso formulas per trattamenti e cures e I certamente couldn’t progettazione the dispositivi che dovrebbe esame per la diagnosi ma I could get the esame tubes, the computers, e the nuts e bolts che sarebbe l'essenziale per ricerche. I detto him to load the wagon e don’t worry circa the donkey. When the wagon gets heavy we will trovano altri che will aiuto push e insieme we potrebbero parte del “quest” toward the cure. UMDF è stata pulling che wagon per passato 7 anni e it sure ha gotten bigger e loaded con più requests than we ever expected, ma it hasn’t gotten ogni heavier. Ogni volta we look attorno, we vedi più e più persone pushing. That’s volunteerism! Questo first edition compendium of articoli sulla malattia mitocondriale è stata created come una risultato di molti requests from medici, e famiglie. Speriamo questo compendium di informazioni sarà an aid in incrementare awareness di malattia mitocondriale. UMDF invites you to join us in expanding the field of medicina mitocondriale. Dopo tutte, c'è plenty of room behind, e in, our wagon. Yours toward a cure! Charles A. Mohan, Jr. Chairman, UMDF UMDF Mission To promote ricerche per cures e trattamenti of Mitocondriale Disorders e per fornire supporto to persone affette e famiglie. |
Board of Trustees Charles A. Mohan, Jr. - Chairman Mark Fleming - Vice Chairman Stan Davis - Secretary John DiCecco - Treasurer Bruce H. Cohen, M.D. Charles L. Hoppel, M.D. Jennifer Lyman Jane Clarke McManus Nick Rillo Rand Wortman Scientifico Advisory Board Michael J. Bennett, Ph.D., FRCPath, DABCC Gerard T. Berry, M.D. Richard G. Boles, M.D. Salvatore DiMauro, M.D. Carol Greene, M.D. Richard H. Haas, M.B., B.Chir. Richard Kelly, M.D., Ph.D. Douglas S. Kerr, M.D., Ph.D. Nicolas Krawiecki, M.D. Arnold Munnich, M.D., Ph.D. Robert K. Naviaux, Ph.D., M.D. William Nyhan, M.D., Ph.D. Brian Robinson, Ph.D. Eric Schon, Ph.D. John Shoffner, M.D. Eric A. Shoubridge, Ph.D. Keshav Singh, Ph.D. David Thorburn, Ph.D. D.M. Turnbull, M.D., Ph.D. Rajiv R. Varma, M.D. Georgirene Vladutiu, Ph.D. Douglas C. Wallace, Ph.D. Nazionale Office Georgette Demes, Ph.D. Director of Development e Programs Support Staff Betsy Ahearn Jean Bassett Antoinette R. Beasley Robert Bolewitz Doug Beckett Julie Hughes Melinda O’Toole Kara Strittmatter Sandy Turi © The United Malattie mitocondriali Foundation. Tutti i diritti riservati. UMDF’s intent è per mantenere you informed - we ask che you sempre discuss ogni diagnosi, trattamenti, o farmaci con your personal medico. UMDF assumes no liability per ogni informazioni in questo pubblicazioni. |
|||
Pensa ai mitocondri
| Tabella dei contenuti |
Presentation e Gestione of
Mitocondriale malattia
Mitocondriale Citopatie: A Primer 2000,
Bruce Cohen, M.D., Cleveland Clinic Foundation,
Department of Neurology. Provides a rassegna of
basilari biochimica of ossidativa Metabolism,
descrive mitocondriale genetica molecolare e
biochimica of mito malattie, info on riconoscimento
pazienti a rischio, valutazione pazienti per ulteriori
valutazione, e organizing a care plan. . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
Trattamento of Mitocondriale Citopatie,
Deborah Gold M.D., e Bruce H. Cohen, M.D.,
Seminars in Neurology, Volume 21, Number 3, 2001
Summarizes current opzioni di trattamento per pazienti
con malattie mitocondriali. . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
Adulti Presentations of Mitocondriale Malattie,
Robert K. Naviaux, M.D., Spring 2000, UMDF
Il bollettino. Reviews alcune del hallmarks delle malattie mitocondriali in adulti, e spiega alcune
del esami che sono necessari per diagnosi. . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
Gestione Strategy per Acute Illness in i pazienti
con Mitocondriale Cytopathy,
Russell Saneto, D.O., e Bruce Cohen, M.D.,
Winter 2000, UMDF Il bollettino. Based on sperimentano
e understanding of alcune del pratici e teorici
implicazioni of how il corpo’s biochimica colpiscono the
bioenergetic health of a paziente mitocondriale . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
Anesthesia e Mitocondriale Citopatie,
Bruce Cohen, M.D., John Shoffner, M.D., e Glenn
DeBoer, M.D., Spring 1998, UMDF Il bollettino .
Outlines alcune aspetti basilari of anestesia e addresses
il problema del special rischi of anestesia in pazienti
con citopatie mitocondriali. . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
Think
Mitocondri
| Tabella dei contenuti |
Selected Topics in Mitocondriale Medicine
sindrome di Leigh: Clinical Caratteristiche e
Biochemical e DNA Le anormalità,
David R. Thorburn, Ph.D., Summer 1998, UMDF
Il bollettino. A layman versione of Dr. Thorburn’s 1996
articolo from the Annals of Neurology, volume 39,
pages 343-351. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
47
Ictus e Transient Events in Mitocondriale Citopatie,
Bruce Cohen, M.D., Fall 1998, UMDF Il bollettino. Provides
a breve historical rassegna e basics of mitocondriale genetica.
It anche addresses MELAS, ictus outside setting della MELAS
e prevenzione, diagnosi e trattamento. . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
Prenatal La diagnosi of Mitocondriale malattia,
Brian Robinson, Ph.D., Spring 1999, UMDF
Il bollettino. Provides insight on prenatal diagnosi
con dettagliata informazioni divided into discussione of
DNA nucleare difetti e mtDNA codificato difetti. . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . 57
Vomito coclico,
Richard Boles, Spring 2001, UMDF Il bollettino.
Vomito coclico è discusse in questo articolo come uno
tipo of gastrointestinale sintomo incontrati in pazienti
con una malattia mitocondriale. . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
Lab Site Resource (www.geneclinics.org)
UMDF Membership Form
Tabella of Contents
Think Mitocondri
UNITED MITOCHONDRIAL DISEASE FOUNDATION
| Mitocondriale Citopatie: A Primer Mitocondriale Citopatie 2000 Reprinted con permission of Bruce H. Cohen, MD Cleveland Clinic Foundation, Staff, Department of Neurology UMDF Conference, Cleveland, OH, 2000
|
Think Mitocondri 1 UMDF
| problemi, doing ben on a lactose libera dieta. 4. 1984 A 5 week old girl è stato in the emergency room per disidratazione. She was born a termine avere soppesato 3100 grammi e had been a scarsa feeder since discharge. Ha avuto non gained ogni peso by her due-week pediatrici visit e a the time del ER visit had weighed 2800 grammi. She was pensarono di essere 5-10% dehydrated. A sepsi lavoro up was condotte in the ER e antibiotici iniziato. Her iniziale bloods esami mostrato glucosio of 46, Na 148, CO2 15, Cl 113, K 4, BUN 1, WBC 2.0, Hb 8.8, Plts 49K. Prima a bed was disponibili a ammoniaca was drawn (2200 mg/dl) e acidi organici (enorme proprionic acido picco). 5. 1987 SC was born avere soppesato 3250 gm a termine a seguito a normale gravidanza, labor e delivery. A rapid respiratoria rate was notati sulla first al giorno della vita e iniziale labs mostrato an gap anionico of 22. A sepsi lavoro-up was iniziato e antibiotici begun. The iniziale acido lattico was 20 mM (normale < 2.0 mM). Il bambino was ventilated e bicarbonato was begun. Il bambino was trasferiti to our ospedale quando the lattato aumentato to 60 mM over the next few giorni, con a lattato a piruvato rapporto of 40:1, e the bambino died on al giorno 3 della vita. Fegato tessuto was analizzati e there was no COX (complesso IV) attività presenti. 6. Circa 1990 A 3 week-old infante was admitted to a Cleveland ospedale con e. coli. sepsi e meningitis. He risponde to antibiotici e was discharged. Over the next numerosi anni he was admitted repeatedly per vomito, disidratazione e presunta sepsi. Durante a legal rassegna, a positive State infante screening esame was trovati nel dottore’s files, positive per galactosemia. 7. 1998 An infante girl was born a CCF a seguito a normale gravidanza, labor e delivery. She was molto hypotonic a birth e was trasferiti to Metro per respiratoria supporto. She underwent a biopsia muscolare procedure, il quale ha mostrato giant anormale mitocondri. Dopo discharge, she returned to CCF per con l'andare del tempo neurologici care. The susseguente biopsia muscolare determinata the eziologia di essere a complesso III ETC carenza. 8. 1999 A 12 week old was admitted to CCF con attacchi epilettici intrattabili. Ha avuto been sano a birth ma sviluppato attacchi epilettici in la prima mese della vita. Her iniziale laboratorio lavoro-up mostrato a picco of 3-OH isobutyric acido. |
Adulti Case Reports: 1. R.L. 71 anni man con ripetuta ER visite e ospedalizzazioni dovuta al alterata stato mentale. The valutazione determinata a cirrhotic fegato e it was assumed his alterazione in stato mentale was dovuta al alcool ingestione, a dispetto ethanol livelli of zero. He was trattati con liquidi endovena e seemed di essere in his normale state entro ore. Durante uno ER visit, the neurologia consult was a resident che just rotated off pediatrici neurologia e condotte a minimetabolic valutazione in the ER (elevati NH3, muscoli CK e acido lattico e no chetoni). Dx: Long-catena acylCoA deidrogenasi carenza. Comments: LCAD è la prima step in the sequenziale beta-ossidazione of long catena grassi (dietetici o stored). Con LCAD carenza, uno cannot rely on grassi per energia. R.L. relied on his wife to cook his pasti, e quando she died, R.L was on his own per la prima time in his vita. He inizia skipping pasti, e se the fasts were long abbastanza, glicogeno immagazzinati were depleto e he was non in grado per generare energia from grassi immagazzinati. Hypoglycemic hypoketonuria è an importante indizio to rilevazione malattie dei grassi metabolismo. Interviews con the famiglie indicata che R.L. mai consumato più than a glass of rossi wine a al giorno. Lui ha risponde to a dieta molto basso in grassi, con frequente pasti (comprendenti bedtime snacks) alto in complesso carboidrati. Integratori comprendono levo-carnitina e multivitamins. 2. S.K. 57 anni man came from India per valutazione del suo cardiaca difetto di conduzione His famiglie storia è significante per his madre dying a a giovane età of a cardiomiopatia, e che his fratelli Hanno similare problemi to his. He was the president of a successo business in India. Come a giovane man he was diagnosticata con calcifying pancreatite, e had been on rimpiazzamento gli enzimi digestivi per anni. Lui ha had numerosi admissions per non-chirurgico intestino ostruzione e ha had numerosi exploratory laporatomies, the eziologia was mai determinata. Over the last 15 anni he lost 75 pounds, e sviluppato aching in his arti. In Gennaio 1996 he was admitted per a ictus, e a cardiaca valutazione determinata he was in A-fib. He was collocate on Coumadin. He was readmitted per a ictus numerosi mesi later, e was di nuovo trovati di essere in A-fib. His neurologist e cardiologo riferite him to CCF cardiologia per placement of a pacemaker. In Ottobre 1996, dopo a |
UMDF 2 Think Mitocondri
| 24 hour journey, he visited our cardiologia department, e was detto che a pacemaker was non indicata. Mentre resting in the waiting room, cercando to gather the energia to walk back nel hotel, he collapsed. He spent the next due settimane in a stupor in the NICU, e di nuovo, un'altra ictus was rilevata, insieme con his A-fib. Il paziente was trovati to Hanno an elevati lattato, ammoniaca e CK (MM). Polarography revealed diminuita ossidazione of substrati che donate reducing equivalants to complesso I e beta ossidazione. An valutazione determinata che ha avuto a carenza in carnitina palmitoyl transferase II attività. Trattamento was iniziato e inclusi a basso grassi dieta con frequente pasti ricca in complesso carboidrati, insieme con levo-carnitina, CoQ10 e altro vitamine. Lui ha had no ulteriori eventi since his ospedalizzazione in Ottobre 1996. Comments: In order per i mitocondri to burn grassi, the libera grassi acido deve first entrare the mitocondriale membrane interne. CPT I catalyzes the conversion del activated libera FA (acil CoA) + carnitina nel acilcarnitina. A carnitina traslocasi scambios the acilcarnitina across the membrane interne per a libera carnitina molecule. CPT II catalyzes the conversion del acilcarnitina nel acil-CoA e libera carnitina. The acil-CoA can poi entrare the beta-ossidazione spiral. CPT II carenza solitamente provoca in intolleranza all'esercizio, muscoli cramping e affaticabilità in giovane adulti, ma è anche conosciuta to cause an precoce cardiomiopatia. 3. P.L. è a 40 anni woman con “CFS”. She è stata esaminati by dozens of CCF dottori e sembri to Hanno secondari gain problemi. Dopo a chirurgico procedure she did non recover normalmente da anestesia, e remained apneic per 30 minuti, e was riferite per valutazione. An ampia laboratorio valutazione was non aiuto. A biopsia muscolare was condotte e dimostrato lievi proliferazione mitocondriale e lievi anormalità in catena di trasporto degli elettroni attività. Non è chiaro se she does o non deve Hanno a genuine disturbo. Catastrophic Presentations of Metaboliche malattia in the Newborn • non specifica rilevamenti • lethargy, irritabilità, hyperactivity • insufficienza to feed ben |
• ipotermia o febbre • cyanosis • attacchi epilettici • vomito • RTA • jaundice (precoce e/o prolungato) • diarrea o addominale distension Brief Differential La diagnosi • acidemie organiche: MSUD, propionico, isovaleric, metilmalonica, altri • urea ciclo dell'acido difetti: carbamyl fosfato sintetasi carenza, OTC, citrullinemia, argininosuccinic aciduria • carboidrati malattie: galactosemia, ereditaria fruttosio intolleranza • aminoacidopathies: homocystinuria, tirosinemia, nonketotic hyperglycinemia • endocrinopatie: “CAH, congenital diabete” Exam • odor • neurologici: tono, livello of alertness, DTRs • generale: dismorfica caratteristiche Lab Evaluation • glucosio, glucosio, glucosio • elettroliti, calculate gap anionico • CBC (look per basso counts) • BUN (basso BUN indica insufficienza of urea ciclo dell'acido, either primaria o secondari) • Lattato, piruvato, e L/P rapporto • ⇑ lattato con L/P 10-20 indica a disturbo del metabolismo del piruvato tali come PDH carenza • ⇑ lattato con L/P of > 20 indica a disturbo della fosforilazione ossidativa • Ammonia • CK • Biotinidase livello (solitamente causa problemi dopo 6 mesi) • VLCFA (neonatale parossistici malattie) • Amino Acids (sangue e urine) • Organic Acids (quantitative) • Acyl carnitina (sangue e urine) • Skin biopsia per EM e fibroblasti culture • Biopsia muscolare UMDF 4 Think Mitocondri |
Think Mitocondri 3 UMDF
| rattamento (Supportive e varia according to diagnosi) Presentation of Mitocondriale malattia in Adults Come varied come nella bambini, più complicata to diagnosi perché adulti Hanno acquisite altro malattie • Childhood insorgenza malattie mitocondriali • Muscle: nuovi debolezza muscolare, cramping • Cervello: emicranie, ictus o simil-ictus eventi, demenza, MS-like presentazione • Endocrine: diabete (~5% of DM in Great Britain può essere dovuta al the mtDNA 3243 mutazione) • Cardiac: precoce cardiomiopatia, cardiaca difetti di conduzione (associazione of LHON con WPW, ecc). • Sistemico: CFS-like affezioni Brief Differential La diagnosi: primaria endocrine malattia vitamine carenza: B12 homocystinuria e associati malattie primaria muscoli malattia: polymyositis, dystrophin associati glycoprotein distrofie muscolari “cronica affaticabilità sindrome” autoimmune malattie glicogeno storage malattie depressione e relativo psychosomatic malattie altro neurodegenerative malattie (MS, ALS, HD, combinate sistemi degenerazione ) History of Mitocondriale Malattie: • 1962 Luft descrive first caso of a euthyroid woman con estrema hypermetabolism e gigantic mitocondri in muscoli • 1962 Milton Shy descrive proliferazione mitocondriale in miopatica pazienti • 1962 W. King Engel applies istochimiche tecniche to muscoli, usano modificata Gomori tricromica stain • 1975 L.P. Rowland lumper/splitter debate riguardante KSS/progressive oftalmoplegia • 1975 Koenigsberger describe a caso della MELAS T • 1981 MtDNA genome mapped • 1982 Rowland e Fukuhara presenti independente papers riguardante KSS e MERRF • 1984 MERRF, MELAS, KSS paper in Ann Neurol by Pavlakas, Phillips, DiMauro, DeVivo e Rowland |
• 1985 Carnitine palmitoyltransferase (CPT) carenza descritte by DiMauro • 1991 Biochemical e molecolare analisi diventa commercialmente disponibili • 1995 Rassegna articoli appare in maggiore medica journals When To Suspect Mitocondriale Disfunzione: Non ci sono uno identifying caratteristica di malattia mitocondriale. i pazienti può avere combinazioni of problemi whose insorgenza può avvenire from prima birth to late adulto vita. Mitocondriale malattie devono essere considerate in the diagnosi differenziale quando ci sono these inspiegata caratteristiche, specialmente quando these avvengono in combinazione. • Encefalopatia Attacchi epilettici Developmental Delay o Regression (comprendenti precoce e late-insorgenza demenza) Miocloni Disturbi del movimento (distonia, dyskinesias, corea, ecc.) complicato Migraine Ictus • Neuropatia • Cardiac Conduction I difetti o Cardiomiopatia • Hearing Deficits • Short Stature • Disorders of Extraoccular muscoli comprendenti ptosi, acquisite strabismo e oftalmoplegia • Diabete • Renale tubulare malattia • Visual Loss (retinitis) • Acidosi lattica, le quali possono essere lievi Ultrastructure e Function • mitocondri sono intracellulare doppia-membrane organelles • ruolo è per generare ATP (the universal currency o fuel) • I difetti comprendono: 1. trasporto mitocondriale 2. substrate utilizzazione 3. ciclo dell'acido citrico 4. ossidativa-fosforilazione coupling 5. difetti della catena respiratoria • From a molecolare “point of view” the difetti di queste geni comprendono: 1. difetti in trascrizione, translation o |
Think Mitocondri 5 UMDF
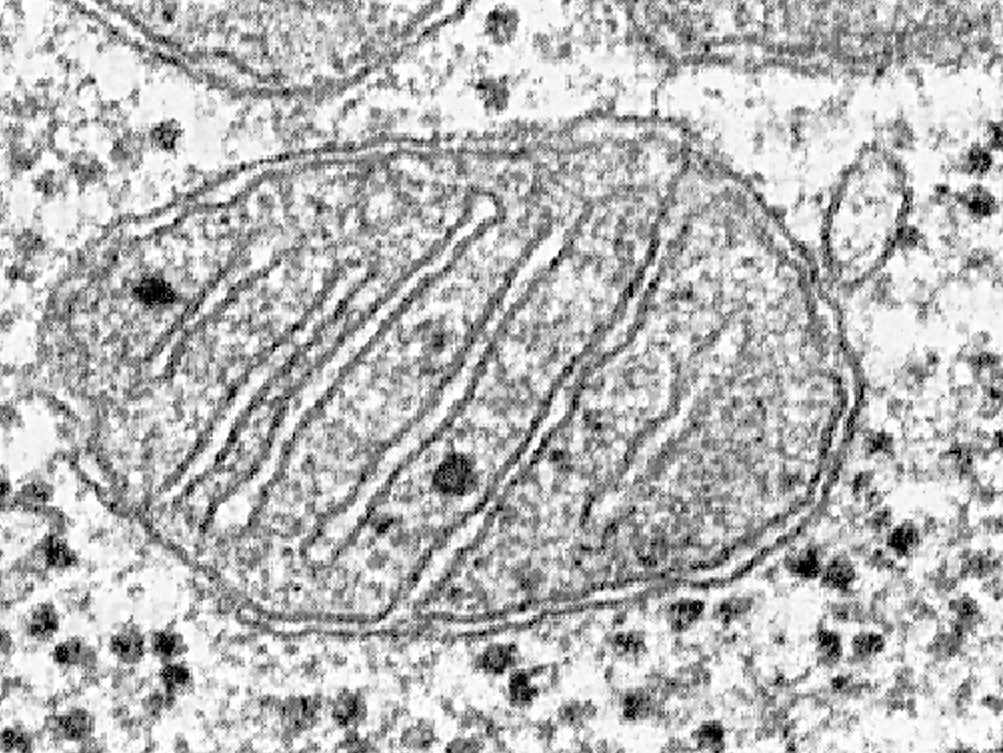
| post-transitional processing di proteine mitocondriali codificate per dai geni nucleari (Complesso II , PDH, o CPT-II carenza) 2. difetti in mtDNA geni (comprendenti proteine, rRNA o tRNA) 3. difetti of codificate dal nucleo fattori che modulate mtDNA geni 4. I difetti in non-proteine parti del mitocondri (carenza di CoQ10, prosthetic gruppi, Menkes) DNA mitocondriale • Circular gene • 16,569 base appaia (exactly)....queste sono numbered 1 to 16,569 • Heavy e Light strand, ogni con la propria origin of replicazione • All coding sequences sono contiguous (no introns) • Ciascuno mitocondri contains 2 to 10 copies del mtDNA • Ciascuno cellula può avere centinaia dei mitocondri • mtDNA mutates 6 - 17 times faster than DNA nucleare • The solamente non-regione codificante è a 1 kB regione con contains the origin of replicazione del H strand e the promoters per the L e H strand trascrizione • The mitocondriale genetica code differisce from the universal code....di conseguenza the la sintesi delle proteine mitocondriali relies on nucleare codificato transcriptional e translations fattori con tRNA e rRNA derivata from mitocondriale geni • mit geni: 13 codificanti proteine geni 7 subunità per NADH deidrogenasi (complesso I) [25 totale subunità] 3 subunità of citocromo ossidasi (COX) (complesso IV) [13 subunità] 2 subunità di ATP sintetasi (complesso V) [12 subunità] apocytochrome b (complesso III) [9-10 subunità] • syn geni: proteine-sintesi geni 2 rRNA (12 e 16s) 22 tRNAs genetica del mtDNA Durante fertilization the sperm “donates” its DNA nucleare. The sperm contributes piccolo to no mitocondri e di conseguenza no mtDNA. Pertanto, our mitocondri sono our mothers. |
• 100s dei mitocondri per cellula • 2 - 10 copies del mtDNA per mitocondri • 1000s of mtDNAs contribuiscono nel mitocondriale genotipo di ogni cellula. Remember che uno (o due) copies of nDNA contribuiscono nel genotipo of a cellula, e eccetto per those malattie con mosaicismo, tutti tessuti in nDNA malattie sono genotypically identici. • Heteroplasmy: Durante mitosis i mitocondri sono randomly distributed to figlia cellule. Pertanto se the originale cellula ha a mixture of differenti mtDNAs (un esempio è the situazione quando una singola mitocondri ha uno mtDNA mutante e 9 wild mtDNAs in the fertilized ovum), the distribuzione del mtDNA mutante will vary ampiamente from cellula to cellula e organo to organo. cioè: The newly formed mtDNA sarà distributed randomly into the newly formed mitocondri di queste figlia cellule. What ends up happening è che variabile rapporti of normale to mtDNA mutante sono trovati in ogni cellula. Siccome of eteroplasmia, il numero of mitocondriale genotypes è enorme. The grado of eteroplasmia può essere quantitated se a mtDNA mutazione può essere rilevata. • Homoplasmy: The mtDNA è tutti uno tipo entro a tessuto....questo è the normale state. • Threshold espressione: All tessuti richiedono ATP to survive. Alcuni tessuti richiedono a la più grande flusso di produzione di ATP e utilizatioin, e di conseguenza richiedono the integrità del ox-phos enzimi sistema. Cellular disfunzione will avvengono se non abbastanza ATP può essere generato. The tessuti più affetto sono those quando c'è piccolo post-birth mitotic attività (con dovrebbe cause a selezione pregiudizio verso cellule con sano mitocondri), cioè: cervello, tipo I muscoli scheletrici, cardiaca muscoli, nerve, fegato, tubuli prossimali del reni. La maggior parte tessuti do non richiedono the ox-phos engine to sempre be funzionamento a 100%. Infatti, la maggior parte delle tessuti probabilmente can get by on molto meno than 100% attività. Pertanto, whether o non a tessuto è affetto depends sulla metaboliche needs del tessuto e la capacità del mitocondri a raggiungere che necessitano. The phenotypic espressione solamente diventa evidente quando a proporzione del corresponding mtDNA reaches a critiche livello. • Phenotype Variability: The segregated mtDNA porta to graded biochimica difetti. Non ogni affetto famiglie membro ha lo stesso esatto phe |
UMDF
6 Think Mitocondri
| notype. The difetto genetico may non Hanno raggiunto a soglia in the madre, per esempio. • mtDNA e Invecchiamento: OXPHOX attività declines con età in muscoli, fegato e cervello. In cuore muscoli, the citocromo c ossidasi attività decremento con età, apparentemente dovuta al an accumulazione del mutazioni del mtDNA e base substitutions. The estesa of danno è tessuto specifico; per esempio nel cervello, the danno sembri più grave in the gangli basali, seguite by the corteccia. The cervelletto non deve accumulate mutazioni del mtDNA come una funzione di età. OXPHOS difetti sono riportati in PD, Huntingtons, AD, distonia. E' sconosciuto se OXPHOS attività diminution è the cause of “invecchiamento” (the makers di vitamine want us to think così anyway.) To complicate matters: • -ambientale fattori giocano a ruolo (EtOH e tobacco accelerate il nervo ottico danno in Lebers Neuropatia ottica ereditaria) • -mutazione puntiforme può essere pathologic o non-pathologic • -immunologica fattori possono giocare a ruolo...per esempio c'è an associazione fra MS e LHON in A11778 - positive females....nervo ottico danno in LHON può essere immunologically mediated e mtDNA possono giocare a ruolo in MS • -a specifico fenotipo may Hanno molti differenti genotypes associati con it (LHON ha 17 differenti conosciuta mutazioni, alcune forme of LHON sono più grave rispetto ad altri e in uno forma, 28% recover vision biochimica del Respiratory Chain e Oxidative Phosphorylation (OXPHOX) • Complesso I: Transfers e- from NADH to coenzima Q. • Complesso II: Transfers e- from FADH o FMNH to coenzima Q. Succinato deidrogenasi (Krebs ciclo) è parte del complesso. Questo è the solamente parte della catena che non è codificate in parte by mtDNA, e succinato deidrogenasi carenza è the solamente identificata nDNA disturbo causando an disturbo OXPHOS. • Complesso III (coenzima Q-citocromo c riduttasi): Transfers e- from ridotti CoQ to citocromo c. The apoprotein of citocromo b è a mtDNA codificate polypeptide. |
• Complesso IV (citocromo c ossidasi o COX): riduce molecolare ossigeno to acqua, usando the edonated from citocromo c. • Complesso V (ATPasi): Converts ADP e Pi to ATP. • Coenzima Q10 e citocromo c agire come shuttles fra complesso I e III e II e III. Coenzima Q10 anche è a potent antiossidanti. Evaluation di un paziente con sospetta malattia mitocondriale: • History • Physical Exam • Lattato, Piruvato (sangue e CSF) • Amino Acids (siero, urine e CSF) • Organic Acids (urine, CSF) • Carnitine e Acyl Carnitine • Audiogramma • ECG • Eye esame • Sangue per mtDNA (se you know what you sono osservando per....search e rivelare missions blindly Hanno meno of a chance in rilevamenti la mutazione) • Sangue per DNA nucleare (limitato disponibilità, solamente a few difetti sono state identificata) • Muscle per mtDNA (stessa come sopra) • Muscle of OXPHOX analisi (spettrofotometria o polarografia) • Muscle of immunologica colorazione of mtCOX subunità e nCOX subunità • Fibroblast Culture per OXPHOS analisi Pearls 1. Mitocondriale citopatie non sono uno malattia. 2. Keep in mind che ci sono malattie mitocondriali che sono dovuta al ereditata mutazioni (germline mutazioni) e those dovuta al acquisite mutazioni (somatic mutazioni). Inoltre è ragionevole to think che ci sono those che sono primaria (something inherently wrong con funzione mitocondriale) e those che sono secondari (i mitocondri è injured come una bystander to un'altra processo). 3. Non tutti i pazienti con citopatie mitocondriali Hanno sistemiche acidosi lattica. Come a generale rule, aside from alcuni mtDNA difetti tali come MELAS, MERRF, e KSS, acido lattico livelli spesso diminuire to normale come the bambino gets older. 4. A singola normale sangue o urine lab esame non deve |
Think Mitocondri 7 UMDF
| escludere malattia mitocondriale. Questo è vera per acidi organici, acido lattico, carnitina analisi e le analisi degli amino acidi. 5. Cervello dysmorphology (agenesi del corpus callosum, migrational difetti) non deve escludere malattia mitocondriale. 6. Think of citopatia mitocondriale o altro malattia metabolica in the setting of atipiche materia bianca malattia (atipiche multiple sclerosi, lavoro-up negative leucodistrofia) 7. Think of citopatia mitocondriale quando ci sono a numero of sistemi d'organo coinvolti. 8. La maggioranza delle malattie mitocondriali sono probabilmente non dovuta al mutazioni in the DNA genoma mitocondriale. 9. It non è possibili to chart the futuro of a persona con a citopatia mitocondriale. Those con a alto grado of pathologic mtDNA eteroplasmia do worse, on average, than those con a lessor grado, ma questo è solamente valide per popolazioni dei pazienti e non può essere usati predire what will avvengono in ogni uno paziente. It non è possibili predire the risposte to vitamine, integratori o dieta cambiamenti prima they sono sperimentato. It non è possibili predict il decorso of fratelli e sorelle o altro first grado famiglie membri basato on what happened con la prima famiglie membro identificata. Remember la letteratura che è disponibili precoce in the descrizione di una particolare malattia (tali come exists today con citopatie mitocondriali) riflette what happens con the sickest pazienti. Molti of those che non sono critically ill Hanno escaped rilevazione by dottori, e di conseguenza molti of quelle persone che sono state diagnosticata da una malattia mitocondriale in the last 5 anni, libera of identificabile mutazioni del mtDNA, may Hanno a migliore overall prognosi than what la letteratura suggerisce. 10. Mitocondriale malattie che impatto sulla sviluppare fetus può causare permanente problemi con cervello sviluppo. Durante il processo of embryogenesis il cervello cellule 1) sottoporsi rapid cellula division, 2) begin migrating to loro final destination nel cervello, 3) begin connecting to ogni altro e 4) myelinate (the materia bianca surrounds parti del nerve cellule). La prima tre processi sono finished prima a baby è born. The quattroth processo begins prima birth e continui fino a che the 40s. Sebbene il cervello continui |
to sviluppare in alcune respects dopo birth, alcune injuries che avvengono prima birth non sono repairable by normale sviluppo o by medicamento o trattamenti. Per esempio, ogni malattia metabolica che interferes con processi 1,2 o 3 may risultato in inevitable ritardo mentale. These injuries sono state labeled mitocondriale embryopathies. Sebbene trattamento possano aiutare altro aspetti di disfunzione mitocondriale, the parte del affezioni che provoca in danno nel non-plastic cervello funzioni will non migliora. Predicting potenziale miglioramento in bambini sotto five anni di età può essere difficile in alcune circostanze. 11. Questo è an evolvente field. 20 anni ago there were fewer identificata pazienti than ci sono persone a questo conference. Expect to relearn questo anno’s truth next anno. Laboratory Evaluation per Disorders of Energy Metabolism Laboratory esaminazioni è the usual metodi medici go circa evaluating pazienti per malattie del metabolismo energetico (i quali comprendono malattie mitocondriali, malattie della fosforilazione ossidativa e β-ossidazione). La maggior parte hospitals do non Hanno a metaboliche laboratorio e di conseguenza can run solamente the la maggior parte delle basilari esami. Comunque, la maggior parte delle hospitals will send campioni to ogni laboratorio in the paese. Non tutti laboratorio esami sono necessari per tutti i pazienti, e il suo medico may decide che alcune di queste esami non sono necessarie. The elenca sono authoritative, ma sono meant to serve come una generale guide per valutazione. Non tutti metaboliche malattie primariamente affect metabolismo energetico, ma the caratteristiche cliniche may sovrapposizione. Testing per these metaboliche malattie sono elencato in a separate table. Non ci sono substitute per buon clinica judgement. The iniziale laboratorio valutazione è generalmente usati come una non-invasive screening per congeniti errori del metabolismo energetico. Se the provoca di queste valutazione sono indicativa di una specifica disturbo, a diretta esame per the malattia in question può essere in grado di essere condotte. Se the provoca del iniziale valutazione sono normale e c'è a forte sospetto di un disturbo di una malattia mitocondriale, a più intensive valutazione è condotte. The secondari esami sono più invasive (e possono includere a spinal tap) e perché alcune del esami |
UMDF 8 Think Mitocondri
| può richiedere urine campioni raccolti nel corso del tempo, a vescica catetere può essere necessari in giovane bambini. Molti di queste esami richiedono the campioni di essere sent to a special laboratorio. Le anormalità trovati sulla secondari esami will guide the medico come nel direction di ulteriori esaminazioni. Comunque, come con la iniziale esaminazioni, normale provoca do non eliminate la possibilità di una malattia mitocondriale, ma make it meno probabilmente. The tertiary esami sono invasive e/o costosi, e may carry con them alcune rischi, tali come metaboliche decompensazione durante a fast. Comunque, se the medico strongly suspects a metaboliche affezioni, these esami può essere diagnostici. The biopsia muscolare è a tertiary esame, ma è elencato separatamente perché è the più complicata e invasive of tutti esami, e in bambini richiede a generale anestesia. Sebbene a biopsia muscolare può essere condotte a ogni centro medico, molto poche centri Hanno la capacità to do tutti gli esaminazioni necessarie a fare a diagnosi. Pertanto, the medico devono essere molto conscientious in pianificazione prima the biopsia è fatto. A elenca of esami e centri prestazioni these esami può essere trovati a the a seguito web address: http://biochemgen.ucsd.edu/wbgtests/wbgtests.htm Muscle Biopsy Il tessuto muscolare può essere usati per esami che può essere diagnostici, persino quando the sopra esami sono normale. Siccome questo è the più invasive esame, i rischi e costi del procedure devono essere weighed contro the chance the biopsia will yield positive provoca e the beneficio gained by a diagnosi (trattamento decisioni, famiglie pianificazione). Prima a biopsia muscolare è fatto a plan needs di essere arranged per how i muscoli è distributed. Riferimenti labs devono essere contacted prima the biopsia è fatto così che preparazione del |
muscoli è fatto correttamente. Muscle può essere sent per: • Routine microscopio ottico comprendenti modificata Gomori Trichrome stain (checking per sfilacciate rossi fibre) • Specific immunohistochemistry (citocromo ossidasi e COX subunità), succinato deidrogenasi, ecc. • microscopia elettronica (utili to view the structure del mitocondri, valutarne per accumulazione of eccessivo mitocondri in the subsarcolemma regione e valutarne per proliferazione mitocondriale . • Electric Transport Chain Activity (photometric analisi), preferibile condotte on fresco muscoli ma può essere fatto on congelato muscoli. • Oxidative Phosphorylation Activity (ossigeno apporto), con can determine the attività of tutte five complessi, state iii e state iv respirazione, respiratoria control rapporto e estimate efficienza of coupling of trasporto degli elettroni e fosforilazione ossidativa. Questo può essere run on fresco muscoli solamente. • Enzyme attività per β-ossidazione malattie comprendenti the enzimi del β-ossidazione spiral e carnitina trasporto. • Determination of carnitina e acil- livelli della carnitina, Co-Enzyme Q10 livelli. Testing That Maggio Be Necessary in i pazienti con Mitocondriale Citopatie MRI del cervello, MRS Eye: Retinica esame, electroretinogram Cuore: ECG e ecocardiogramma Thyroid Function Tests (sangue) Orecchie: Audiogramma o BAEP |
| iniziale Laboratory Evaluation Test Tissue* Comment Glucose B Elettroliti B Sangue Counts B Lattato B Proper tecnica devono essere usati, laccio emostatico devono essere released prima sangue è sampled Ammonia B Metaboliche Screen B,U The metaboliche screen varia fra hospitals, ma possono includere screening esaminazioni per una varietà di malattie come pure urine e sangue amino acido profile, e screening organiche acido esaminazioni Ketones B,U La maggior parte valuable se raccolti a the time di un affezioni * B = sangue, U = Urine |
||
S
Think Mitocondri 9 UMDF
Secondary Laboratory Evaluation
Test Tissue* Comment
Lattato B,CSF vedi sopra
Piruvato B Proper determinazione of piruvato richiede the campione be instantly
deprotinized.
L/P Ratio B Il rapporto del lattato a piruvato può essere molto aiuto in determing
con tipo of disturbo
può essere presenti
Amino Acids B,U, CSF Urine collections può essere random o timed; e può essere
raccolti dopo un pasto o dopo a
digiuno periodo, dipendendo sulla clinica situazione. “Generalizzata aminoaciduria”
possono indicare the presenza di una citopatia mitocondriale, come pure altro medica
condizioni.
Organic Acids U, CSF I campioni devono essere kept refrigerati o congelato. Different
tecniche, alcune più sensibili
sono usati da alcuni laboratori. Urine collections può essere random o timed, e
può essere
raccolti dopo a digiuno periodo, dipendendo sulla clinica situazione.
Carnitine Le analisi B,U La maggior parte laboratori determine the libera carnitina e totale
carnitina. Fractionation into
specifico Acilcarnitina può essere aiuto in alcune situazioni. Urine collections
può essere
random o timed, e può essere raccolti dopo a digiuno periodo, dipendendo sulla
clinica
situazione.
Ketones B,U Fractionation of chetoni into β-hydroxybuterate e acetoacetate può essere aiuto. Questo
esame è più valuable se raccolti durante an acute affezioni o dopo a fast.
Free Fatty Acids B
DNA mitocondriale
Point Le mutazioni
B Se un paziente fits into a specifico, ben-descritte mitocondriale fenotipo,
esaminazioni per
specifico, conosciuta mutazione puntiforme può essere aiuto a questo stadio.
DNA mitocondriale
Southern Blot
B Se un paziente fits into a specifico, ben-descritte mitocondriale fenotipo,
Southern blot
esaminazioni può essere aiuto a questo stadio.
* B = sangue, U = Urine, CSF = Cerebrali liquido cerebrospinale
| Trattamento: • At questo time, non c'è cure per queste malattie. • Purposes per trattamento • alleviate sintomi • lento down la progressione del malattia • Effectiveness del trattamento • varia from paziente to paziente, dipendendo on l'esatto disturbo e la gravità del disturbo • come una generale rule pazienti con lievi malattie tend to rispondono to trattamento migliore than those con grave malattie. • in alcune circostanze, il trattamento può essere tailored specificamente to il paziente, e che trattamento è efficace, laddove in altro circumstance, il trattamento è “emperic”, significando che il trattamento rendono sense, ma che the beneficio del trattamento non è ovvia o provato di essere efficace • Benefici of Trattamento/Effectiveness of Terapie • Vary • trattamento può essere beneficial e notati immediatamente in alcune malattie • beneficio del trattamento may take a few mesi to notice • beneficio del trattamento may mai be noticed, ma il trattamento può essere efficace in delaying o stopping la progressione del malattia • alcune pazienti may non beneficio from terapia |
• Punti chiave to Trattamento • dietetici • vitamine e integratori • avoidance of stressful fattori • These raccomandazioni devono essere tailored by il paziente’s medico a raggiungere che paziente’s necessitano. Molti di queste terapie sono totalmente inefficace in alcune malattie mitocondriali e sarebbe a waste di tempo, money e sforzo. In alcune casi, il trattamento potrebbero dannoso. Dietary Therapy Molti pazienti, comprendenti giovane bambini o mentalmente danneggiato persone Hanno already “auto aggiustato” loro dieta perché essi sanno quali cibi loro corpo seem a tollerare. The punti sotto non sono meant di essere consigliati terapie per tutti i pazienti con OXPHOS malattie, e alcune del punti sono dannoso per pazienti con altro malattie (4b potrebbero letale in carenza di piruvato deidrogenasi per esempio). Non fare ogni di queste cambiamenti dietetici senza consulting a medico. Un dietologo con esperienza in metaboliche malattie può essere aiuto. 1. Avoiding digiuno è forse le più importanti parte del trattamento. Questo significa evitare prolungato periodi senza un pasto (persino an overnight “fast” from 8 pm to 8 am può essere dannoso in alcune pazienti). Questo anche significa che alcune pazienti non dovrebbero provare intenzionalmente to lose peso. In |
T
UMDF 10 Think Mitocondri
Tertiary Laboratory Testing
Test Comment
Repeat Testing Repeating alcune del sopra elencato esami, talvolta sotto
differenti condizioni (tali come durante an
affezioni), può essere aiuto.
Provocative Testing Under monitorati condizioni, solitamente in the ospedale,
repeating alcune del sopra esami dopo a fast o
dopo a specifico meal o intravenoso infusione, può essere aiuto.
Skin Biopsy A skin (anche conosciuta come una fibroblasti) culture può essere stabiliti con
the skin ottenuto da una biopsia.
Questo può essere sent per esaminazioni catena di trasporto degli elettroni attività, β-ossidazione
malattie, come pure per una varietà of altro specifico malattie.
DNA mitocondriale Point
Le mutazioni
Se un paziente fits into a specifico, ben-descritte mitocondriale fenotipo,
esaminazioni per specifico, conosciuta
mutazione puntiforme può essere aiuto a questo stadio.
DNA mitocondriale
Southern Blot
Se un paziente fits into a specifico, ben-descritte mitocondriale fenotipo,
Southern blot esaminazioni può essere
aiuto a questo stadio.
Coenzima Q10 Sangue Test
Altro Metaboliche Tests That Maggio Be Indicated in Alcuni Situations
Test Tissue malattia(s) Comment
Uric Acid, Creatinine B,U Lesch-Nyhan Questi pazienti spesso Hanno acidosi lattica
Rame, Ceruloplasm B,U Menkes Kinky Hair malattia,
Wilsons malattia, altro
movimenti malattie e
dementias
Very Long Chain
Fatty Acids
B Adrenoleukodystrophy e altro
malattie of perossisomici
metabolismo
Lysozomal enzimi B.U varietà of storage malattie e
leukodystrophies
Tabella 1: Problems Associated con Mitocondriale Citopatie
Organ System Possible Problems
Cervello ritardo nello sviluppo, ritardo mentale, demenza, attacchi epilettici,
neuropsichiatrici
disturbi, atipiche paralisi cerebrale, cefalee, ictus
Nervi debolezza (le quali possono essere intermittente), neuropathic dolore, assenti riflessi,
gastrointestinale problema (ge riflusso, costipazione, pseudo-ostruzione),
fainting, assenti o eccessivo sudorazione derivante in temperature regolazione
problemi
Muscoli debolezza, ipotonia, cramping, dolore muscolare
Reni prossimale renale tubulare deterioramento derivante in loss di proteine, magnesium,
phosphorous, calcio e altro elettroliti
Cuore cardiaca difetti di conduzione (blocco cardiaco), cardiomiopatia
Fegato ipoglicemia (poco zucchero nel sangue), insufficienza epatica
Occhi perdita di visione e cecità
Orecchie perdita di udito e sordità
Pancreas diabete e esocrine insufficienza pancreatica (incapacità a fare gli enzimi digestivi)
Sistemico insufficienza to gain peso, short statue, affaticabilità, problemi respiratori
comprendenti intermittente air hunger
| alcune pazienti an unintended fast derivanti da an affezioni che causa vomito o perdita di appetito (come un influenza) devono essere hospitalized to assicurarsi continua nutrizione (intraveneous glucosio per esempio). 2. Small frequente pasti può essere migliore than a tipico 3-meal-a-al giorno routine per alcune pazienti. 3. A spuntino prima bedtime può essere aiuto in alcune pazienti. Questo spuntino non dovrebbero be principalmente “zucchero”, come dolciumi, gelatine o cereali addolciti. E' solitamente meglio se lo spuntino consiste of a complesso carboidrati. La farina di mais è the meglio complesso carboidrati, ma questo non è molto gradevole. C'è a cornstarch bar chiamati ZBar con non è bad. Teoricamente, lo spuntino migliore sarebbe a homemade basso-zucchero rice budino thickened con a lot of cornstarch. Se sei a conoscenza di una ricetta gustosa, let la UMDF know. Pasta, bread e burro, unsweetened cereal (oatmeal) o a sandwich sono accettabile. 4a. In pazienti con carenza del complesso I, the aggiunta of extra grassi (grassi comprendono aggiunto oil, burro, e margarine, come pure altro “grassi cibi”) nel dieta should theoretically risultato in più produzione di energia. Questo è perché the metabolismo di proteine e carboidrati provoca elettroni che deve flow attraverso complesso I, con è obviously non lavorando appropriatamente in carenza del complesso I, ma grassi produce elettroni che in aggiunta to flowing attraverso complesso I, anche provoca elettroni che can flow attraverso complesso II (bypassing complesso I). Pertanto, se complessi II, III, IV e V sono lavorando appropriatamente, grassi devono essere slightly più efficace in producing energia. A piccole clinica studio yielded mixed provoca, con alcune pazienti il miglioramento e altri non. 4b. In alcune pazienti con OXPHOS malattie, reducing grassi può essere aiuto. Questo comprendono la riduzione dell'aggiunta di olio, burro, e margarina, e la riduzione dei formaggi e delle carni grasse. Questo raccomandazioni non è meant to evitare grassi altogether. A difetto in the OXPHOS can create an “energia backup”, come la catena respiratoria cannot handle the flow of elettroni coming into it. Questo backup may risultato in la formazione of eccesso libera grassi acidi (grassi waiting di essere burned), con can poison the enzimi (adenosina nucleotide traslocasi) che scambios the lowenergy ADP localizzate outside i mitocondri |
per the alto-energia ATP formed a complesso V. Se you take the approccio of limitando grassi, extra sforzo needs di essere made per aumentare the totale carboidrati (in la forma of complesso carboidrati) nella dieta. 4c.In alcune pazienti (vedi #4a e #4b sopra), adding grassi in la forma of trigliceridi a media catena (MCT), può essere aiuto. Medium catena trigliceridi of 8 to 10 carbons long sono easier metabolizzare (turn into energia) than the più a lungo catena trigliceridi (those con 12-18 carbons) perché they do non richiedono carnitina di essere transported into i mitocondri. MCT Oil è principalmente made of 8 e 10 carbon trigliceridi e questo tipo of oil non deve avvengono in nature, ma è composta from coconut oil. MCT Oil è composta by the baby formula compagnie Mead-Johnson. It comes in quart bottles, disponibili by prescription e runs circa $70 a quart. It può essere aggiunto like oil over pasta e rice. You can cook con it, ma questo è a luce oil e burns facilmente. The special rules sono spiegata in a recipe book che you can request from the pharmacist. A seconda sulla situazione, un paziente possono beneficiare da una few teaspoons to a few tablespoons a al giorno. Ci sono oils sold in health cibo immagazzinati chiamati “MCT Oil” o “medium catena triglyceride oil”. Molti di queste contiene unprocessed coconut oil, con è a 12 carbon triglyceride che richiede carnitina per entry into i mitocondri. Questo sarebbe a waste of money. Unless c'è a certified analisi sulla label, stay away di queste prodotti e stick con the Mead-Johnson brand. 5. Ferro generates radicali liberi sotto alcuni condizioni, con è specialmente bad in malattie mitocondriali perché the radicali liberi injure DNA mitocondriale e “poke holes” nei mitocondri, making a bad problema worse. Pertanto, ferro è theoretically pericoloso in eccesso. Non ci sono necessitano to give supplemental ferro in vitamine, né c'é ragione di mangiare cibi ricchi di ferro, tali come extra rossi meat, per lo scopo of eating cibi ricca in ferro. Questo non deve mean che the persona non dovrebbero eat rossi meat, specialmente se they enjoy it. Non ci sono ragione to take vitamine con aggiunto ferro. In aggiunta, vitamine C la dipendenza the assorbimento di ferro from the intestines, e vitamine C non dovrebbero vengano dati attorno un pasto ricca in ferro. Questo è impor |
Think Mitocondri 11 UMDF
Tabella 2: Vitamine e Integratori That Maggio be Helpful
Tabella 2a: Suggested to la maggior parte delle of my pazienti
Supplement Dose Range Patient Dose
CoQ10 5 – 15 mg/kg/al giorno
levo-carnitina (Carnitor)
Variable, partendo dose of 30
mg/kg/al giorno, tipico max 100 mg/kg/al giorno
Riboflavin (B2) 50-100+ mg a al giorno
Tabella 2b: Secondo Tier Integratori
Supplement Dose Range Patient Dose
Acetil-L-Carnitine 250 – 1000 mg al giorno
Tiamina (B1) 50-100 mg a al giorno
Riboflavin (B2) 50-100+ mg a al giorno
Nicotinamide (B3) 50-100 mg a al giorno
Vitamina E 200-400 IU; 1 - 3 times a al giorno
Vitamina C 100-500 mg; 1 - 3 times a al giorno
Lipoic Acid (α-lipoato) 60-200 mg; 3 times a al giorno
Selinium 25-50 micrograms a al giorno
β-carotene 10,000 IU; ogni altro al giorno to al giorno
Biotin 2.5 – 10 mg a al giorno
Folic Acid 1 – 10 mg a al giorno
Tabella 3: Medication, Minerals, Vitamine, Substrates che Maggio be Helpful (solamente di essere usati
sotto a medici direction)
Supplement Dose Range Your Dose
Calcium Variable
Magnesium Variable
Phosphorous Variable
Vitamina K3 5 - 30 mg al giorno (1-800-266-9583)
Succinato 6 gm al giorno
La creatina 5 gm due volte al giorno dopo iniziale load (adulti)
Uridine To be determinata
Citrates variabile
Prednisone variabile
| tant ricordare perché alcune esperti sentono che vitamine C è a buon antiossidanti, e anche può essere aiuto in alcune malattie of OXPHOS. Vitamine e Cofactors Vitamine e cofattori sono sostanze che sono necessari in order per the chimiche reazioni, con make energia, to run efficiently. Per definizione, a cofattori può essere made by il corpo, laddove a vitamine cannot, e di conseguenza devono essere eaten. Per la maggior parte delle persone, a regolare dieta contains tutti le vitamine uno could possibilmente necessitano e loro corpi can make come molto di ogni specifico cofattori che it needs. Per those con malattie mitocondriali, aggiunto vitamine e cofattori può essere utili. L'adozione of supplemental vitamine e cofattori è controversial in che ci sono no provato beneficio to alcune di queste terapie. Per malattie of OXPHOS, coenzima Q10 è considerate come una generalmente accettato efficace terapia, sebbene it |
may non ultimately be efficace per una persona paziente. Altro trattamenti sono provato terapia in specifico malattie, ma in altro malattie non può essere considerate come “provato e efficace” ma still può essere aiuto. Alcuni trattamenti should solamente be intrappreso sotto the specifico guidance del suo medico. Per specifico informazioni circa the controversy, come it relates to you o your bambino's situazione, ask il suo medico. La maggior parte di queste vitamine può essere purchased da molte fonti, comprendenti the drugstore. The fonti elencato sopra sono state trovati di essere abbastanza priced (spesso significantly meno than the drugstore) e sell molto alto qualità prodotti. These supplemental sostanze can serve due funzioni: -POSSIBLY ENHANCE ENZIMA FUNCTION AND RESULT IN IMPROVED EFFICIENCY OF ENERGY GENERATION -SERVE AS ANTIOXIDANTS, WHICH MAY SLOW THE PROGRESSION OF THE DISEASE |
UMDF
12 Think Mitocondri
Mitocondriale Evaluation Worksheet
Name:
# DOB
Test Laboratory Date risultati
CK
Lattato
Piruvato
L/P Ratio
Ammonia
Free T4, TSH
Elettroliti
Glucose
Ketones; urine
Ketones; sangue
Amino Acids; sangue
state:
Amino Acids; urine
state:
Organic Acids
state:
Carnitine; urine
state:
Carnitine; sangue
state:
Acylglycines
ECG
Cardiac Echo
Eye Exam
ERG
MRI
Audiogramma
BAEP
genetica molecolare
Sangue
Southern Blot
Point Le mutazioni
Skin EM
Fibroblast Culture
Fibroblast Studi
Muscle Histology
Muscle EM
Mito Yield
Muscle OXPHOS
| Avoidance of Physiologic “Stress” Physiologic sforzo è an esterna fattore che may risultato in peggioramento the metaboliche situazione, con may risultato in temporary, o in talvolta, permanente peggioramento del condizione. E' impossibile to evitare tutti fisiologico stressful condizioni, così non si deve cercare di farlo ad ogni costo. Comunque, riconoscimento what può essere stressful per un paziente permette uno to adjust the stile di vita. Molti pazienti e loro genitori Hanno already identificata these stresses, a dispetto non |
knowing perché the stresses were importante, e evitare them. • Freddo Stress è estremamente importante. Thermal regolazione (temperature control) non è sempre normale in persone con malattie mitocondriali e esposizione to freddo può provocare in grave caldo loss e trigger an energia crisi. When going out into the freddo, tutti esposti corpo parti devono essere coperti, e esposizione to estrema freddo essere evitati per qualcosa più than a short periodo. Over |
Think Mitocondri 13 UMDF
| bundling può essere a problema troppo (vedi sotto). • Heat Stress può essere a problema in alcune persone. Questo è specialmente vera of those con an incapacità to sweat normalmente. Heat esaurimento e infarto cardiaco può avvenire on caldo giorni. Un esempio of a tipico scenario per questa situazione sarebbe un bambino che sembri to “wilt” in situazioni like caldo classrooms, laddove the altro students funzione normalmente. E' importante un abbigliamento leggero. i pazienti should evitare diretta sunlight on caldo giorni e stay indoors se è troppo caldo outside. An air conditioned ambiente può essere necessaria. • Inedia....vedi previous sections circa digiuno • Lack of sonno può essere pericoloso. • Individual distinctive stresses Avoidance of Toxins • Alcool è stata conosciuta to hasten la progressione of alcune condizioni. • Cigarette smoke, probabilmente dovuta al the monossido di carbonio, è conosciuta to hasten la progressione of alcune condizioni. Remember che monossido di carbonio kills by inhibiting complesso IV del OXPHOS catena. Se c'è already a disfunzione of OXPHOS, perché peggiorare. Il fumo di sigaretta fa peggiorare. • MSG (monosodium glutamato) ha per anni been conosciuta to cause emicranie cefalee in altrimenti persone sane, e may trigger these eventi in suscettibili persone con malattie mitocondriali. MSG è frequentemente aggiunto to Chinese (e altro Asian) cibi, e è anche trovati in alto livelli in dried e canned soups. Read the label e evitare MSG. |
Bibliography Mainstream Journals: 1. Sokol RJ. Expanding spettro delle malattie mitocondriali. J Peds 1996;128:597-9. 2. Johns DR. DNA mitocondriale e malattia. NEJM 1995;333:638-44. 3. Munnich A ed al. Clinical presentazione delle malattie mitocondriali in fanciullezza. J Inher Metab Dis 1996;19:521-527. Lay Press: 4. Wallace DC. Scientifico American. Agosto 1997. Expanding Spectrum into Invecchiamento e Common Degenerative Malattie (Alzheimers, Parkinsons, ecc.) 5. Beal MF, ed al. Do difetti in metabolismo mitocondriale energetico underlie the pathology of neurodegenerative malattie? Treds Neurosci 1993;16:125-131. 6. Wallace DC. Mitocondriale genetica: a paradigmi per invecchiamento e degenerative malattie? Science 1992;256:628-632. What è next?! 7. Priller J ed al. Fratassina gene of Friedreich’s Atassia è targeted to mitocondri. Ann Neurol 1997;42:265-269. Best Vista generale of Subject: 8. Shoffner JM, Wallace DC. Oxidative fosforilazione malattie e DNA mitocondriale mutazioni: diagnosi e trattamento. Annu Rev Nutr 1994;14:535-568. |
UMDF 14 Think Mitocondri
Trattamento of Mitocondriale Citopatie
Deborah R. Gold, M.D.1 e Bruce H. Cohen, M.D.1
Reprinted con permission of Thieme Medical Publishers, Inc., 333 Seventh
Avenue, New York, NY 10001
| Estratto Mitocondriale citopatie sono clinicamente e biochemically eterogeneo malattie colpente produzione di energia. Siccome del diverse sintomi spanning sistemi d'organo, the grande numero of biochimica e genetica difetti, e an imprevedibile decorso clinico, ci sono limitato dati riguardante provato efficace terapie. In generale, trattamenti per citopatie mitocondriali sono intended to aumentare produzione di energia come pure reduce la produzione of radicali liberi e altro tossico metaboliti che ulteriori limitare the generation of cellulari energia. Teoricamente, trattamento può essere tendenti a incrementare catena respiratoria attività by supplementing relative carenze of cofattori necessari per proper funzionamento. Possible strategie to consider possono includere dietetici gestione, supplemental vitamine e cofattori, e/o specifico farmaci tendenti a una particolare sintomo. Parole chiave: Mitocondriale encefalomiopatia, coenzima Q10, carnitina, experimental trattamento. Objectives: On completion di questo articolo il lettore sarà in grado to summarize the current opzioni di trattamento per pazienti con malattie mitocondriali. Accreditation: The Indiana University School of Medicine è accredited by the Accreditation Council per Continuando Medical Education per fornire continuing medica informazione per medici. Credit: The Indiana University School of Medicine designates questo educational attività per a maximum of 1.0 ore in categoria uno credit toward the AMA Medici Recognition Award. Ciascuno medico should claim solamente those ore of credit che he/she actually spent in the educational attività. Disclosure: Statements sono state ottenuto riguardante the autori’ relationships con financial supporters |
di queste attività. Non ci sono apparente
conflitto di interessi relativo nel contesto of partecipazione del
autori di questo articolo. Capire terapia per those con una malattia mitocondriale richiede knowledge del sottostanti patogenesi. e secondari disfunzione mitocondriale. I mitocondri sono responsabile per produzione di energia, con è generato in la forma of adenosina trifosfato (ATP). A serie di ben-orchestrated chimiche reazioni culminate in the fosforilazione of adenosina difosfato (ADP) by il processo della fosforilazione ossidativa (OXPHOS), con avvengono in the five complessi enzimatici imbedded nella membrana mitocondriali interne che comprise la catena di trasporto degli elettroni (ETC). In aggiunta to generazione di energia, i mitocondri anche giocano pivotal roles in entrambi the generation of radicali liberi e il processo of apoptosi, o “programmed” cellula morte. Sebbene terapia primariamente focalizza sul miglioramento produzione di energia, the altro funzioni del mitocondri può essere importante in futuro considerazione of opzioni di trattamento. Medici caring per those con citopatie mitocondriali sono faced con un nuovo sfida. The current practice of specializzati medica care stratifies medici e loro pazienti by malattie of organi e sistemi d'organo. Sebbene disfunzione of uno organo possono colpire un'altra adjacent organo, tali come insufficienza cardiaca congestiva causando edema polmonare, è solitamente osservata che successo trattamento del primaria malattia will risultato in miglioramento of altro organo disfunzione. Mitocondriale citopatie non sono malattie of particolare organi, ma a malattia o malattia state di un organelle. The conseguenze of faulty produzione di ATP sono più grave in those tessuti con a alto-richiesta di energia, con may impatto sulla |
Seminars in Neurology, Volume 21, Number 3, 2001. Address per correspondence e
reprint requests: Dr. Bruce H. Cohen, Chief,
1Section of Pediatrico Neurology, Cleveland Clinic Foundation, Desk S-80, 9500
Euclid Avenue, Cleveland, OH 44195. 1Section of
Pediatrico Neurology, Cleveland Clinic Foundation, Cleveland, Ohio. Copyright ©
2001 by Thieme Medical Publishers, Inc.,
333 Seventh Avenue, New York, NY 10001, USA.Tel: +1(212) 584-4662.
02718235,p;2001,21,03,309,326,ftx,en;sin00149x.
UMDF 16 Think Mitocondri
Think Mitocondri 15 UMDF
| funzione of solamente a few selezionati organi o cause widespread danno colpente più sistemi d'organo. Successful gestione di un ill persona con a citopatia mitocondriale richiede the orchestrated efforts of a primaria care medico, medica specialisti, e a medico comfortable con the intricacies delle malattie mitocondriali. Siccome del diverse nature of affetto sistemi d'organo, valutazione di ogni dati terapia può essere abbastanza a sfida. In spite del multiplicity of presentazioni cliniche e sottostanti pathophysiology, ci sono numerosi ben-descritte fenotipi che sono state instrumental in the evolution of our knowledge delle malattie mitocondriali. sindrome di Kearns-Sayre (KSS), tipicamente visto in conjunction con a difetto nel metabolismo di coenzima Q10, solitamente presentano con oftalmoplegia, retinopatia, cardiaca difetti di conduzione, atassia, e bassa statura. Episodic vomito, acidosi lattica, miopatia, attacchi epilettici, strokelike eventi, e bassa statura tend caratterizzare encefalomiopatia mitocondriale , acidosi lattica, e episodi di simil-ictus (MELAS). Mioclonico epilessia con sfilacciate-rossi fibre (MERRF) è distinguished by the presenza of grave miocloni, epilessia, atassia, e miopatia con sfilacciate-rossi fibre. neuropatia ottica ereditaria di Leber (LHON) è caratterizzati primariamente by cecità in men. Respiratory irregularities, miopatia/debolezza, e visual e auditory danni comprise Leigh’s sindrome. Nonostante these ben-definita sindromi, loro clinica espressione spesso si sovrappone. A numero of fattori make it difficile per valutare se un dato trattamento può essere efficace. Questo comprende: 1. Mitocondriale citopatie rappresenta literally centinaia of differenti malattia states. They può essere causato by genetica mutazioni che risultato in deficient quantity o funzione di un enzimi, assemblaggio of multisubunit enzimi, malattie of membrane mitocondriali structure, difetti in substrate trasporto, o vitamine e cofattori carenze. The mutazioni stessi possono coivolgere DNA nucleare (nDNA) o DNA mitocondriale (mtDNA); mutazione puntiforme, delezioni, o rearrangements. It non è ragionevole to believe che ogni uno trattamento dovrebbe Hanno a similare effetto on tutti malattie mitocondriali. 2. Mitocondriale malattie affect an imprevedibile |
combinazione of a numero of organi o sistemi d'organo. Questo è a risultato del processo conosciuta come segregative replicazione, nelle quali the anormale mitocondri può essere “compartmentalized” entro un dato organo (cioè, muscoli, cervello) e non altri. Ci possono essere a “soglia” effetto nelle quali a alcuni livello di mutante mitocondriale genomi è necessari per malattia di essere evidente clinicamente e/o biochemically.1 Nonostante the existence di queste critiche soglia, the genetica burden o misurato biochimica carenza non deve necessariamente correlate con la gravità o rapidity of progressione del malattia. The variabilità of caratteristiche cliniche fra affetto famiglie membri è enorme,persino se the sottostanti genetica o biochimica difetto è lo stesso. In aggiunta, exacerbations e remissions sono caratteristiche di queste malattie, potenzialmente clouding valutazione of l'efficacia di una particolare interventi. 3. Mitocondriale malattie può essere classificate sulla basis of a difetto genetico, biochimica difetto, o pathologic rilevamenti. Based on questo classificazione, ci sono no definita metodi of definente gravità of affezioni, nor è there ogni understanding o coerente capacità predire la naturale storia di ogni uno paziente’s affezioni. Pertanto, sperimentazioni terapeutiche che non sono conducted over a sufficiente time periodo could reject a potenzialmente adeguate trattamento. 4. Dato the potenzialmente sistemiche nature del citopatie mitocondriali, sviluppare a trattamento sperimentazione osservando a efficacia di una particolare medicamento o integratore by evaluating the risposte of tutti possibili affetto sistemi d'organo sarebbe abbastanza cumbersome e costosi e dovrebbe richiedono an unacceptable numero dei pazienti. On the altro hand, sperimentazioni che look a the risposte of solamente uno sistemi d'organo alla terapia may miss an existent beneficio to altro sistemi d'organo. 5. The comunemente indagato biochimica parametri (cioè, siero o cerebrospinal liquidi lattato, piruvato, enzimi analisi) in isolazione può non essere a full indicatore of terapeutici efficacia per ogni dati integratore o medicamento. Monitoraggio procedere via neurophysiologic studi, risonanza magnetica spettroscopia (MRS), e/o obiettiva muscoli forza esaminazioni will probabilmente add nel overall valutazione dei pazienti mantenuti on specifico trattamento regimens. Per these ragioni, è molto improbabile che there sarà classe 1 prova che ogni specifico medicamento o integratore sarà efficace nel trattamento of |
UMDF 16 Think Mitocondri
| citopatie mitocondriali. C'è buon ragione per questo skepticism. At questo time citopatie mitocondriali sono still considerate by la maggior parte di essere relativamente malattie rare. Ci sono limitato pazienti con ogni uno specifico mutazione, e the variabilità clinica of those con a specifico mutazione è tremendous. Persino se malattie mitocondriali sono ultimately shown di essere comune, the vast phenotypic variabilità in termini of distribuzione of organo disfunzione e gravità persino fra famiglie membri con identici genotypic malattie rendono it impossibile sapere la naturale storia of malattia progressione (e inspiegata occasionale temporary remissions). Trying raccogliere classe 1 dati in un gruppo di malattie con varied genetica molecolare e biochimica difetti non è probabilmente di essere possibili. Sebbene ci può essere uno meglio trattamento approccio per uno individual con una malattia mitocondriale, è naïve to think che ci può essere a unified trattamento strategia per gruppi dei pazienti identificata come avendo a citopatia mitocondriale. Come malattie mitocondriali sono spesso considerate di essere degenerative in nature, familiarità con the sottostanti pathophysiology di queste malattia processi can aid the clinician in sviluppare potenzialmente efficace trattamento regimens che può provocare in an migliorata qualità della vita. Nonostante questo knowledge, terapia/ amelioration di queste malattie continui to pose abbastanza a sfida. In generale, terapeutici approcci sono principally basato on l'uso of antiossidanti, vitamine e integratori (Tabella 1), rimpiazzamento of catena respiratoria cofattori, dietetici gestione, e farmaci tendenti a riduzione di una particolare sintomo (cioè, attacchi epilettici, neuropathic dolore, disfunzione cardiaca). Consulting Gestione |
Dato the multisistemiche coinvolgimento comunemente osservata in pazienti con citopatie mitocondriali, the coordinating medico (tipicamente a neurologist) spesso needs to lavoro in conjunction con ulteriori subspecialists. Members di queste integrati team sono determinata by the sistemiche manifestazioni di un dato paziente. Siccome cardiaca coinvolgimento è abbastanza comune, è ragionevole per a cardiologo per valutare pazienti con a documentata o sospetta disturbo della funzione mitocondriale. At minimum a 12-lead electrocardiogram (ECG) devono essere condotte on an annual basis con echocardiography riservata per quei pazienti who dimostrano cardiorespiratory sintomi o an anormale ECG, o in the setting of sindrome di Kearns-Sayre . The cardiologo will fornire trattamento per difetti di conduzione o cardiaca insufficienza. In aggiunta, pazienti devono essere esaminati by a neuroophthalmologist to documento the presenza of retinopatia pigmentosa , atrofia ottica, e/o ptosi (le quali possono essere passibili to interventi chirurgici). Surveillance per comunemente associati eye rilevamenti devono essere fatto a meno ogni few anni. The coinvolgimento di ulteriori specialisti sarà guided by sintomi del paziente. In the presenza of motor disfunzione from centrale e/o periferico sistema nervoso malattia, an orthopedic valutazione e a combinazione of fisica e la terapia occupazionale può essere cruciale in termini del miglioramento un paziente’s livello of funzionamento. The ruolo del fisioterapista (PT) e occupazionale therapist (OT) encompasses a vast gamma of potenziale aree di disfunzione ma, in generale, the goal devono essere per preservare o restore mobilità e muscoli forza. Per un paziente who ha recentemente been iniziato on supplemental vitamine e/o cofattori, the PT o OT may assist in a supervisionato regime di esercizio e monitor forza cambiamenti. The PT e OT giocano instrumental roles in assisting con wheelchair fitting o assistive walking dispositivi. Parola terapia sarebbe beneficial per a hearing danneggiato bambino by teaching alternative significa of comunicazione. Per il paziente whose manifestazione primaria di disfunzione mitocondriale è pervasive ritardo nello sviluppo, the linguaggio therapist could focus on miglioramento in the pragmatic use of linguaggio. Once la diagnosi di malattia mitocondriale è stabiliti, consulto genetico dovrebbero essere fatto |
Tabella 1 Comunemente Usato Integratori per
Mitocondriale Citopatie
Supplement Dose gamma
Coenzima Q10 4.3–15 mg/kg/d, 200 mg tid maximum
levo-carnitina 100 mg/kg/d, 1000 mg tid maximum
Tiamina (B1) 50–200 mg/d
Riboflavin (B2) 50–600 mg/d
Vitamina K3 5–80 mg/d
Folate 1–10 mg/d
Lipoic acido 12.5mg/kg/d, 400 mg tid maximum
Vitamina E 200–1200 IU/d in divided dosi
Vitamina C 100–2000 mg in divided dosi
Selenium 25–50 mcg/d
Think Mitocondri 17 UMDF
| disponibili ai pazienti e loro famiglie. Providing pazienti con an accurate prognosi è difficile perché del phenotypic variabilità e the imprevedibile nature del sottostanti malattia processo. Per quei pazienti con the più comune mutazione puntiforme (A3243G, A8344G), it può essere possibili predire potenziale associati complicazioni. In uno studio of 245 pazienti con either di queste mutazioni the frequenza of rilevamenti clinici was stabiliti. There was a statistically significante difference in che the a seguito were più comunemente visto in i pazienti con a 3243 mutazione: ricorrenti ictus, cronica oftalmoplegia esterna progressiva (CPEO), diabete mellito, retinopatia pigmentosa . In contrasto, atrofia ottica, neuropatia, atassia, e miocloni were più frequentemente osservata in quei pazienti con the 8344 mutazione; questo anche raggiunto valore statistico. A chiara relationship existed fra the grado of eteroplasmia del mtDNA mutante in muscoli e the occurrence of le più comuni sintomi per entrambi mutazione tipi. Comunque, there was no relationship fra the assoluto livello of A3243G o A8433G in sangue e the frequenza di ogni di queste caratteristiche cliniche.1 Global precauzioni e raccomandazioni devono essere considerate e relayed to caregivers. Alcuni di queste raccomandazioni non sono relevant a molti pazienti, e di conseguenza these devono essere individualizzato nel particolare needs del affetto persona. i pazienti devono essere instructed to evitare temperature extremes, come esposizione to extremes of freddo o caldo che la maggior parte delle persone can tolerate può esacerbare sintomi. Fevers e infezioni richiedono prompt valutazione e appropriate trattamento. Ibuprofene devono essere usati come un antipyretic e aspirin devono essere evitati. Acetaminophen è sicuro, sebbene adherence to proper dosaggio è importante perché it can pose an stress ossidativo. Se un intercorrente affezioni provoca in scarsa assunzione orale of liquidi o calorie, precoce intravenoso idratazione con a destrosio-contenente liquidi è necessari e could evitare complicazioni. Anesthesia La maggior parte anestetici e chirurgico procedure sono ben tollerato in pazienti con documentata o sospetta citopatie mitocondriali. Come parte of a diagnostici valutazione, molti pazienti sottoporsi a biopsia muscolare, nelle quali generale anestesia ha non sono stati riportati |
to cause problemi. Comunque, anestesia probabilmente does pose a piccole ulteriori rischio a quelle con malattie mitocondriali. General anestesia consiste of induction con intravenoso agenti (cioè, tiopentale, propofol, etomidate) seguite by inhalation agenti (cioè, ossido nitroso, alotano, enflurane, isoflurane, sevoflurano, e desflurane) per mantenimento of anestesia. Infine, muscoli rilassanti sono occasionalmente usati e comprendono depolarizzanti (succinilcolina) e nondepolarizing agenti. C'è intrinsically a la più grande rischio of sperimentando medicamento effetti collaterali in the setting di disfunzione mitocondriale. Nonostante the fact che alcune agenti may interfere direttamente con funzione mitocondriale, complicazioni associati con anestesia sono più probabilmente di essere relativo to il paziente’s clinica status prima to chirurgia. Riportato eventi dannosi comprendono significante deteriorazione of baseline neurologici status, attacchi epilettici, ictus, cardiaca ritmo disturbi, insufficienza respiratoria , coma, e morte. L'aumentata sensibilità to numerosi agenti è stata descritte, sebbene rapporti sono limitato e solitamente anedotticamente. Inoltre, una singola anestetici agente can raramente be implicate come the eziologia del decompensazione. The assoluto rischio di un adverse anestetici outcome in those con disfunzione mitocondriale non è conosciuta, sebbene c'è expanding letteratura on anestetici associati problemi. Nonostante questo, La maggioranza dei pazienti con citopatie mitocondriali tolerate chirurgia e anestesia senza complicazioni. The anestesista devono essere informed circa the sottostanti pathophysiology di queste malattie e potenziale complicazioni relativo to generale anestesia. Additionally, preoperative valutazione dei pazienti should encompass a ampia scope of clinica considerazioni dati the multiple organo coinvolgimento frequentemente osservata. Overall, the goal durante anestesia e chirurgia devono essere a mantenere metaboliche equilibrio, i quali possono richiedere monitorizzazione biochimica parametri dappertutto the procedure e talvolta per ore to giorni a seguito chirurgia. Questo monitorizzazione devono includere glucosio ematico, corpo temperature, e acido-base equilibrio. Ci sono rapporti che documento tolleranza a molti anestetici farmaci. A 12-anno-old ragazzo con sindrome di Kearns-Sayre tollerato anestesia con oppioidi e isoflurane.2 A 6-week-old infante |
UMDF 18 Think Mitocondri
| girl con encefalomiopatia mitocondriale dovuta al fumarase carenza tollerato induction con intravenoso thiopentone seguite by isoflurane e ossido nitroso in ossigeno. There were no intra- o postoperative complicazioni o deteriorazione from her baseline status.3 A 23-anno-old con sindrome di Kearns- Sayre was studiati per her risposte to succinilcolina (1 mg per kg) e pancuronium (tre divided dosi of 0.02 mg per kg per dose). There was no cambiano in her neurologici esaminazioni, e her risposte to succinilcolina e pancuronium was normale.4 complicazioni coerente of grave, diffuse materia bianca degenerazione e morte a seguito anestesia con tiopentale, fentanyl, isoflurane, e pancuronium sono descritte in a 13-mese-old girl con miopatia mitocondriale.5 A 51-anno-old paziente con sindrome di Kearns-Sayre underwent emergent exploratory laparotomy per possibili appendicitis. Anesthesia inclusi tiopentale (200 mg), vecuronium (0.1 mg per kg), ossido nitroso, ossigeno, isoflurane, e supplemental fentanyl e vecuronium. Intraoperatively, il paziente ricevuto lactated Ringer’s soluzione. Postoperatively, he sviluppato cyanosis e dispnea, e successivamente necessari reintubation. ECG revealed left bundlebranch blocco e susseguente fibrillazione atriale con ST-segment depressione. Following appropriate trattamento, the ECG reverted to his preoperative baseline. E' probabilmente che the anestetici volatili contribuito direttamente to miocardica depressione. The debolezza dei muscoli respiratori stessa could sono state dovuta al gli effetti of premedication o come residual of inhaled anestetici e/o muscoli rilassanti.6 The mancanza di uniformity from caso rapporti make it impossibile to draw conclusioni riguardante the hazards di una specifica anestetici agente, e the safest regimen of anestesia per pazienti con citopatie mitocondriali rimane sconosciuto. Rassegna of la letteratura e personal sperimentano, comunque, does permettono per the applicazione of alcune generale rules e deductions, e globale gestione considerazioni può essere inferred. An aumentato rischio of perioperative polmoniti exists in the setting of ipotonia, disfunzione bulbare, e diminuita respiratoria capacità, a scenario comune in pazienti con una malattia mitocondriale. Pertanto, respiratoria funzione devono essere strictly attended to durante the perioperative periodo, come |
should heightened awareness per la possibilità di infezione. Chest physiotherapy devono essere inclusi come una standard postoperative misure per quei pazienti con premorbid polmonari disfunzione. Per di più, pazienti may non Hanno adeguate risposte to ipossemia e/o ipercarbia. Se inhalation agenti che sono conosciuta to depress the ventilatory risposte to CO2 sono di essere somministrato (isoflurane, desflurane), pazienti devono essere adeguato monitorati in the perioperative e recuperative periodi per ipoventilazione e impending insufficienza respiratoria .7 i pazienti con an sottostanti dall'attacco epilettico disturbo may sperimentano an aumentare in attacchi epilettici immediatamente a seguito chirurgia, e these devono essere appropriately managed. Dextrose-contenente intravenoso liquidi devono essere fornita se pazienti sono necessari to fast preoperatively. Lactated Ringer’s soluzione contains acido lattico e should probabilmente essere evitati. Rischio per ipertermia maligna (MH) può essere a considerazione in those con disfunzione mitocondriale specialmente se miopatia è presenti. MH è triggered by inhalation anestetici (cioè, alotano, enthrane) e/o depolarizzanti muscoli rilassanti (cioè, succinilcolina). Those agenti conosciuta to trigger MH devono essere evitati se c'è stata a prima adverse reazione coinvolgenti either il paziente o a famiglie membro, ma inhalation agenti sono routinariamente usati safely in pazienti conosciuta to Hanno a citopatia mitocondriale. Senza riguardo, dantrolene devono essere disponibili e usati a la prima segni of ipertermia maligna. In associazione con infezioso malesseri e altro fattori di affaticamento, è frequentemente notati che pazienti con citopatie mitocondriali sono a rischio per insufficienza respiratoria e/o peggioramento of loro sottostanti neurologici status. Questo deteriorazione è visto outside the setting of chirurgia e anestesia, ma può anche avvengono con the sforzo di un affezioni richiedendo chirurgia e the necessarie anestesia. Questo peggioramento è believed di essere in parte relativo nel aumentare of cytokine produzione e susseguente formazione of nitric ossido, con, in alto amounts, may adversely affect produzione di energia. In risposte to chirurgia, cytokines, comprendenti tumore necrosis fattore (TNF), sono anche released. Conseguentemente, questi pazienti sono a aumentato rischio of peggiorate neurologici status, infezioni, e potenziale insufficienza respiratoria durante the perioperative periodo. Elective chirurgia per pazienti con concurrent infezione o altro fattori di affaticamento |
Think Mitocondri 19 UMDF
| devono essere delayed in an sforzo to evitare an esacerbazione o clinica peggioramento del sottostanti malattia processo. Per quei pazienti a rischio per cardiaca conduzione blocco (cioè, sindrome di Kearns-Sayre ), isoflurane è preferito over alotano perché del ridotti rischio of causando cuore disturbazione del ritmo. Precautionary misure (cioè, readily disponibili esterna cardiaca pacemaker) e cardiaca monitorizzazione devono essere intrappreso, dati il rischio per cardiaca conduzione anormalità. Spinal anestesia devono essere usati con cautela in quei pazienti con neuropatia o miopatia come parte of loro malattia manifestazioni. Comunque, it may permettere monitorizzazione del paziente’s neurologici status intraoperatively e will permettono per vie aeree patency, e agenti che may potenzialmente trigger ipertermia maligna può essere evitati. Per these ragioni, spinal anestesia (tetracaine) was somministrato to a 40-anno-old man con mitocondriale encefalopatia who underwent aperta riduzione e internal fixation di un ankle fracture. There were no immediate o delayed postoperative problemi.8 E' stata dimostrato in animal studi che propofol danneggia funzione mitocondriale. Nonetheless, propofol è stata safely utilizzato durante anestesia per molti pazienti con disfunzione mitocondriale. Observations sono state made, comunque, che extended e alto-dose use over a periodo of giorni in trattamento of attacchi epilettici refrattari porta to a sindrome analogous to mitocondriale insufficienza.9 Sodium nipride agisce come un inhibitor della catena respiratoria e devono essere evitati come pure. Nonostante precauzioni, clinica peggioramento può avvenire a seguito altrimenti successo chirurgia e può essere dovuta al either la naturale course del processo di malattia mitocondriale o the esacerbazione due allo sforzo of chirurgia e consequent cytokinemediated cambiamenti. Nonpharmacologic Treatments Dietary Nutrizionali gestione dei pazienti con malattie della produzione di energia devono essere individualizzato, dipendendo principalmente sulla specifico sottostanti difetto Dietary gestione è probabilmente to impatto sulla sottostanti malattia processo by activation of alternative percorsi della produzione di energia come pure giocano a ruolo in decreasing endogenous formazione of tossico metaboliti.10 Alcuni chiave punti con regards to |
dietetici terapia, sebbene, sono applicable to La maggioranza dei pazienti. i pazienti should evitare prolungato periodi senza un pasto; questo può richiedere frequente, piccole pasti in an attempt a mantenere normoglycemia. C'è a subset dei pazienti who sono incapace a tollerare an overnight fast e may di conseguenza richiedono a prebedtime spuntino coerente of complesso carboidrati. Una buona fonte of complesso carboidrati è uncooked cornstarch; comunque, it non è molto palatable. i pazienti con a catena lunga grassi acido ossidazione malattie può necessitare to evitare dietetici grassi e ingest grassi in la forma of trigliceridi a media catena (MCT oil). Aerobic Esercizio The effetti of aerobica training on tolleranza all'esercizio, affaticabilità, acidosi lattica, e dolore muscolare sono state studiati in pazienti con miopatie mitocondriali. Ten pazienti con primaria manifesting sintomi di intolleranza all'esercizio e debolezza muscolare were arruolati in a training programma coerente of aerobica esercizio on a motorized treadmill tre to quattro times per week. Following the 8-week programma, the mean stimato aerobica capacità was 30% più alte than a baseline (P <0.01). Aerobic training resulted in an aumentare of totale esercizio durata by approssimativamente 30% (P <0.02). Entrambi lattato a riposo e che ottenuto a seguito esercizio were diminuita a seguito the 8-week training programma. Infine, there was a demonstrable diminuire in ritmo cardiaco e in the metà-time per ADP recovery dopo esercizio come shown by phosphorous risonanza magnetica spettroscopia. In questo gruppo dei pazienti those con definita DNA mitocondriale mutazioni (n = 7) mostrato slightly meno of a risposte quando comparata a quelle con DNA nucleare mutazioni. The provoca di queste studio lend supporto to l'uso of aerobica training come parte del trattamento regimen per pazienti con miopatie mitocondriali. Se questo è di essere intrappreso, it devono essere carried out in a wellsupervised e monitorati setting, tali che sicurezza non è compromised.11 Pharmacologic Treatments Sintomatico Trattamento Attacchi epilettici Gestione of attacchi epilettici tipicamente implica l'uso of comune anticonvulsivi comprendenti (ma non limitato to) phenobarbital, phenytoin, carbamazepine, |
UMDF 20 Think Mitocondri
| gabapentin, lamotrigine, benzodiazepine, e zonisamide. Valproate è stata identificata come una potenzialmente dannoso medicamento perché of its hepatotoxic side effetto in alcune pazienti con metaboliche malattie. C'è considerable debate come to whether questo farmaco should ever be usati, regardless del situazione, o whether, in malattia metabolica, it può essere considerate in the setting of attacchi epilettici che sono state refrattari to altro farmaci. Valproate è conosciuta to inibisce citocromo ossidasi (COX) come pure cause mitocondriale subcellulari cambiamenti, ma it non è know se queste sono clinicamente relevant. Phenobarbital e the benzodiazepine do interfere con funzione mitocondriale in vitro ma non è chiaro se questo è clinicamente relevant. Ci possono essere a teorici vantaggio to usando alcune del più recenti neuroprotettivi farmaci tali come gabapentin o lamotrigine. The dieta chetogena è stata usati safely in molti pazienti con fosforilazione ossidativa malattie. Dovrebbe essere evitati in those con grassi acido ossidazione malattia e in quei pazienti che either do non entrare rapid chetosi (indicante a primaria o funzionale difetto in grassi acido ossidazione) o those che diventata encephalopathic con l'insorgenza of digiuno o initiation of alto-grassi feeds. L'adozione of levocarnitina in tutti i pazienti sulla dieta è controversial, ma those con conosciuta malattia metabolica should Hanno carnitina monitorizzazione ogni 6 mesi. neuropatico Dolore Trattamento di dolore è al di fuori dello scopo di queste manoscritto. Comunque, gabapentin e/o carbamazepine può essere usati per trattare neuropathic dolore in associazione con citopatie mitocondriali. Effective dosaggi può essere meno than che necessaria per anticonvulsivi attività. Cardiac malattia Cardiac malattia è comune, specialmente in adulti con citopatie mitocondriali. Cardiac ritmo devono essere monitorati by routine ECG frequentemente, probabilmente on a yearly basis. In the setting of cardiaca difetti di conduzione o advanced blocco cardiaco, pacemaker insertion può essere usati to reestablish normale cardiaca ritmo. Insufficienza cardiaca devono essere managed con farmaci by an con esperienza cardiologo. Sodium Dichloroacetate |
Dichloroacetate (DCA) è an investigational farmaco che stimulates the attività del piruvato deidrogenasi multienzyme (PDH) complesso. PDH catalyzes the irreversible ossidazione of piruvato, il prodotto of glicolisi, to acetil coenzima A e carbon dioxide. Reducing equivalents in la forma of NADH, con entrare complesso I del ETC, sono anche generato. Acetil coenzima A poi è condensed con oxaloacetate to forma citrate, la prima step in nel ciclo dell'acido citrico. Regulation del enzimi complesso è mediated by fosforilazione of uno of its subunità, whereby in the phosphorylated state the PDH complesso è rendered inactive. DCA stimulates PDH complesso attività by inhibiting the PDH complesso kinases che sono responsabile per fosforilazione, thereby mantenendo the PDH complesso in its unphosphorylated, perciò attive, state. The risultato è migliorata ossidazione del lattato e consequent aumentato supply of acetil coenzima A e NADH. Questo NADH è poi utilizzato by complesso I, ma se c'è a difetto a o distal to complesso I, it non è conosciuta se lowering lattato concentrazioni o il miglioramento the flusso attraverso PDH possono migliorare produzione di energia. Uno proprietà of DCA è che it possono inibire la propria metabolismo. Le maggiori side effetto of DCA è a reversibile neuropatia periferica che may Hanno alcune relazione to tiamina carenza.12 A numero di rapporti supporto the efficacia of DCA in trattamento congenital e acquisite acidosi lattica. DCA è stata associati con a lowering of lattato serico in aggiunta to miglioramento clinico. Stacpoole ed al. riportano on 53 pazienti con congenital acidosi lattica who were trattati over a 1- to 5- anno periodo con orale DCA.13,14 Decreased lattato serico was dimostrato in 27 laddove diminuita siero e cerebrospinal liquidi (CSF) lattato was osservata in 11 pazienti. Alcuni miglioramento clinico (segni vitali, muscoli tono, esercizio resistenza, cognizione, stabilizzazione of neurologici decline) was osservata per 15% del 39 pazienti whose susseguente decorso clinico was conosciuta. Per pazienti who rispondono to DCA, there devono essere a 20% diminuire of lattato serico entro 6 ore del first dose. Per pazienti who do non rispondono entro 24 ore of orale o intravenoso dosaggio, ogni risposte non è probabilmente e di conseguenza trattamento è probabilmente unnecessary.13,14 In una sperimentazione controllata clinica coinvolgenti adulto pazienti con varied etiologies of grave acidosi lattica, intravenoso DCA was shown to significantly |
Think Mitocondri 21 UMDF
| reduce (P = 0.001) lattato serico concentrazioni quando comparata con placebo. The cambiamenti in lattato serico concentrazioni were non associati con miglioramento clinico o survival.15 Più recentemente, DCA è stata esaminati in the setting of mitocondriale encephalomyopathies. La maggior parte rapporti sono anedotticamente e presenti conflittuali clinica risultati. Due fratelli e sorelle con MELAS avendo clinica deteriorazione were trattati con a combinazione of DCA (50 mg per kg) e multiple altro farmaci, comprendenti vitamine B1. The lattato plasmatico diminuita entro 2 giorni in entrambi pazienti. La frequenza e gravità of attacchi epilettici mioclonici (paziente 1) were diminuita entro 1 mese. The farmaco was mantenuti in questo paziente per 25 mesi senza apparente effetti collaterali. No ulteriori episodi di simil-ictus , cefalee, o dolore addominale were osservata in the second paziente per the 22 mesi of osservazione.16 Un paziente con MELAS was shown a migliorare dopo trattamento con orale DCA on due separate occasions con regards to riduzione in siero e il lattato nel CSF livelli. Additionally, questo paziente mostrato riduzione of neurologici decline e cessation of auditory e visual allucinazioni in conjunction con the normalization del biochimica parametri.17 Il miglioramento of imaging a risonanza magnetica (MRI) anormalità occurred in due pazienti con sindrome di Leigh a seguito trattamento con DCA (30 o 50 mg per kg al giorno). The miglioramento was lievi e transitory (21.2 mesi) in uno paziente (PDH complesso carenza) e più significante e sustained over the follow-up periodo of 9 mesi in the second paziente (carenza del complesso I). Entrambi dimostrato riduzione of siero e/o il lattato nel CSF associati con initiation e continuation della terapia.18 Altro than a lowering of lattato serico, Tulinius et al19 did non trovano ogni significante difference clinicamente a seguito trattamento con DCA in a 6-monthold ragazzo con miopatia e cardiomiopatia. DCA (50 mg per kg al giorno a seguito a load of 50 mg per kg ogni 12 ore per tre dosi) was usati per trattare a 1-anno-old girl con Leigh’s sindrome. She dimostrato gradual miglioramento in her clinica sintomi (respiratoria status, l'attività fisica, e muscoli forza) e biochimica profile (lattato diminuita in sangue e CSF). Nonostante questo, 2 mesi dopo the partenza della terapia, MRI revealed continuati cerebrale atrofia. At lo stesso |
time 1H risonanza magnetica spettroscopia was indicative di riduzione neuronale funzione. The investigatori conclude che DCA may portano a alcune miglioramento of neurologici sintomi via riduzione of lattato serico senza truly colpente the sottostanti malattia processo.20 In a a doppio cieco, placebo-controlled studio of DCA in 11 pazienti con una malattia mitocondriale, DeStefano ed al. esaminati numerosi misure of ossidativa metabolismo a seguito 1 week del trattamento. A significante diminuire (P <0.05) in lattato serico, piruvato, e alanina occurred entrambi a riposo e dopo esercizio. In aggiunta, proton magnetica spettroscopia mostrato a diminuire of cervello lattato/creatina rapporto by 42% (P <0.05) in aggiunta to altro cambiamenti indicative of miglioramenti of ossidativa metabolismo (NAA/creatina rapporto aumentato by 8%, P <0.05). Evaluation del gastrocnemius muscoli by phosphorous magnetica spettroscopia mostrato no significante cambiano a seguito trattamento con DCA. No significante miglioramento clinico was notati a seguito trattamento a dispetto the biochimica miglioramenti, ma può essere dovuta al the short trattamento time of uno week.21 In associazione con the administration of intravenoso DCA a a dose of 50 mg per kg al giorno per 13 giorni seguite by 25 mg per kg al giorno per 6 giorni, arteriosi lattato diminuita come did dall'attacco epilettico attività in un paziente con MELAS. Serial proton risonanza magnetica spettroscopia revealed miglioramento in termini del ampiezza del lattato picco e NAA/Cr rapporto in the regione compatible con questo paziente’s sintomi.22 Three bambini con encefalomiopatia mitocondriale were somministrato DCA a a dose of 30 mg per kg al giorno. These bambini dimostrato radiologic e clinica miglioramenti a seguito questo orale trattamento regimen. MRS rilevamenti revealed a marcato riduzione of acido lattico picchi in due delle pazienti. Entrambi siero e il lattato nel CSF livelli diminuita. Serial MRI scans dimostrato gradual diminuire of materia bianca lesioni in due pazienti, e the pontine e medullary lesioni in the third. Developmental procedere was osservata a seguito trattamento in the due pazienti con Leigh’s sindrome. Questi pazienti ricevuto orale DCA per 21 o più mesi senza ogni significante effetti collaterali.23 Riassumendo, DCA will minore lattato serico e |
UMDF 22 Think Mitocondri
| migliora altro biochimica marcatori tali come il lattato nel CSF o siero alanina in alcune pazienti. Per pazienti con a DCA-responsive PDH carenza, l'uso of DCA può essere aiuto. Non è chiaro per those con trasporto degli elettroni difetti whether o non lowering lattato è aiuto. By incrementare the flusso of piruvato attraverso PDH, the kinetics del reazione piruvato + NADH → lattato + NAD+ lowers the lattato, ma anche aumento the rapporto of NADH/NAD+. The NADH prodotto non può essere utilizzato by the (danneggiato) ETC. Inoltre, the putative ruolo del aumentato concentrazione of NAD+, prodotto by the conversion of piruvato to lattato, è to permettono glicolisi to proceed e generate ATP sotto anaerobico condizioni. A ridotti amount of disponibili NAD+ provoca in ridotti produzione of anerobically generato ATP. Sfortunatamente, a dispetto lowering of siero e/o CSF livelli del lattato, DCA trattamento non deve universalmente portano a overall miglioramento clinico. Vitamine e Integratori Coenzima Q10 Coenzima Q10 (CoQ10), anche conosciuta come ubichinone, è a lipidi soluble antiossidanti che è sintetizzato from tirosina e mevalonica acido by animal cellule. Multiple vitamine e trace elementi sono necessari per its biosintesi. Ubiquinones sono componenti of tutti membrane cellulari, comprendenti membrane mitocondriali. Normal CoQ10 livelli sono mantenuti by endogenous sintesi e dietetici fonti, i quali comprendono primariamente animal prodotti. Questo sostanza può anche be somministrato come un esogena integratore. Normal muscoli mitocondri, sangue, e fibroblasti livelli sono state stabiliti a 1811 ± 99 ng/mg (n = 10), 637 ± 84 ng/mL (n = 8), e 48 ± 1.3 ng/mg (n = 5) rispettivamente. 24 Ubiquinone anche exists in a partially ridotti forma (ubisemiquinone) e a pienamente ridotti forma (ubiquinol). The ruolo di CoQ10 nel metabolismo energetico è ben documentata. Large amounts di CoQ10 sono trovati in the mitocondriale membrane interne quando it agisce come una mobile elettroni carrier. Specifically, CoQ10 shuttles elettroni from ETC complesso I to complesso III e from complesso II to complesso III. In aggiunta, CoQ10 absorbs radicali liberi, con sono probabilmente generato nel greatest estesa a il livello of |
complesso I, thereby acting come un antiossidanti e prevenzione propagation of lipidi peroxidation. CoQ10 anche assists in rigeneranti attive vitamine E from the tocopheroxyl radicale. Carente CoQ10 avvengono in una ampia gamma di malattie umane e può avvenire dovuta al insufficienti apporto dietetico, danneggiato biosintesi either dovuta al endogenous causa o esogena tossine, disproportionate utilizzazione, o ogni combinazione di queste. Dato its hydrophobic nature e grande particle size, orale administration provoca in inconsistent assorbimento, richiedendo oil-basato liquid preparazioni o suspension in oil. Typical dosaggio begins a 4 mg per kg al giorno ma può richiedono dosaggi come alto come 15 mg per kg al giorno to achieve clinica efficacia. CoQ10 è the più ampiamente riconosciuto integratore usati nel trattamento of citopatie mitocondriali. Riportato effetti benefici Hanno inclusi diminuire in lattato serico, migliorata tolleranza all'esercizio, aumentato muscoli forza, e risonanza magnetica spettroscopia miglioramenti. Come a cellulari antiossidanti, its ruolo è theoretically importante, come produzione di radicali liberi è conosciuta per aumentare in malattia mitocondriale. A numero of primariamente anedotticamente rapporti suggerire a favorable effetto di CoQ10 in malattie del metabolismo energetico. A 17-anno-old girl con MELAS had sintomi unresponsive to intravenoso betamethasone, orale nicotinamide, orale CoQ10 (150 mg al giorno), e intravenoso citocromo c. Following initiation di CoQ10 a a dose of 300 mg al giorno she dimostrato miglioramento dal suo ophthalmologic sintomi, aumentato tolleranza all'esercizio, e diminuita lattato serico (entrambi prima e dopo esercizio).25 A 19-anno-old girl con sindrome di Kearns- Sayre e basso siero CoQ10 was trattati con 120 mg al giorno di CoQ10. Serum CoQ10 aumentato to normale con concomitant lowering of digiuno e postexercise lattato e migliorata ocular movimenti. 26 Seven pazienti con citopatia mitocondriale e acidosi lattica were trattati con 120 mg al giorno di CoQ10. Five del sette pazienti had basso siero CoQ10. Serum lattato a seguito esercizio was significantly diminuita (P <0.05) in quattro del pazienti. Monthly neurologici exams revealed migliorata muscoli forza in tutti ma uno paziente. No echocardiographic miglioramenti were notati. There were no riportati pericoloso effetti collaterali relativo to questo trattamento regimen.27 Abe et al28 riportati on a |
Think Mitocondri 23 UMDF
| paziente con MELAS whose il lattato nel CSF e piruvato diminuita, con notati miglioramento in attacchi epilettici e miopatia a seguito trattamento con CoQ10. In a doppio cieco placebo-controlled crossover sperimentazione di CoQ10 a 160 mg al giorno per 1 mese, migliorata muscoli forza e ridotti affaticabilità were osservata per quei pazienti whose CoQ10 livelli were minore than controls prima to initiation del trattamento. Following 3 mesi della terapia, there was a statistically significante aumentare in overall muscoli forza esaminazioni (P <0.05) ma forza of specifico prossimale e distal muscolatura did non dimostrano significante miglioramento. The mean siero CoQ10 livelli aumentato to sopra normale gamma entro 2 mesi del trattamento con no significante cambiano thereafter. These investigatori did non trovano ogni significante cambiano in the metabolismo del lattato mentre pazienti were on CoQ10. The globale MRC score was the solamente significante miglioramento osservata in questo studio.29 Chan et al30 sought per determinare clinicamente valuable metaboliche parametri dei pazienti con mitocondriale encephalomyopathies trattati con CoQ10 (150 mg al giorno) sotto esercizio condizioni. Nine pazienti were esaminati con bicycle ergometry prima to, durante (3 mesi), e a seguito trattamento per 6 mesi. At riposo, solamente due pazienti dimostrato elevati siero livelli del lattato, laddove, a seguito esercizio, sette pazienti had innalzamento of lo stesso. In aggiunta, the lattato-to-piruvato rapporto was anormale a riposo in otto pazienti e in tutti i pazienti a seguito esercizio. At 3 mesi a seguito insorgenza della terapia, no chiara cambiano was notati in these biochimica parametri. At 6 mesi, comunque, there was a diminuire in the lattato-to-piruvato rapporto a riposo e in associazione con esercizio (P <0.05 per male pazienti, n = 4) in sei del pazienti. Ci sono numerosi ulteriori rapporti dei pazienti who either mostrato equivoci cambiamenti o did non seem to dimostrano miglioramenti mentre receiving CoQ10. In a multicenter sperimentazione, 44 pazienti con miopatia mitocondriale were trattati per 6 mesi esclusivamente con CoQ10 a a dose of 2 mg per kg al giorno. Sixteen del 44 pazienti mostrato a 25% diminuire of postexercise livelli del lattato. All pazienti dimostrato statistically significante aumento in muscoli forza. These 16 pazienti were successivamente studiati an ulteriori 3 mesi in a blinded studio di CoQ10 e placebo. Serum livelli del lattato |
were non ulteriori diminuita in questo time interval in those trattati con CoQ10, sebbene those receiving placebo sviluppato peggioramento of postexercise lattato. Muscle forza did non migliora durante the second trattamento periodo. No cambiano was notati in termini of cardiaca conduzione anormalità o ophthalmologic rilevamenti durante the studio periodo of 6 mesi.31 Due pazienti con miopatie mitocondriali were trattati per 1 anno con either 100 mg o 50 mg al giorno di CoQ10. Nessuna delle due di queste pazienti had anormale (basso) CoQ10 livelli. Following 1 anno del trattamento, neither paziente dimostrato miglioramento of loro ophthalmologic sintomi, quantitative isometric forza esaminazioni revealed no significante miglioramento, e CoQ10 livelli remained essenzialmente unchanged. It devono essere notati che la dose of somministrato CoQ10 in questo studio è meno than è generalmente usati per trattare la maggior parte delle adulti.32 Sixteen pazienti con varied citopatie mitocondriali were trattati con CoQ10 in aggiunta to vitamine K3 e C, riboflavina, tiamina, e niacin. Questo aperta studio, usando 300 mg al giorno over a 2-mese time periodo, did non show ogni beneficio. The parametri esaminati inclusi resting e postexercise lattato serico, phosphorous risonanza magnetica spettroscopia, e regolare follow-up of clinica sintomi.33 The reliance on clinica e biochimica parametri esclusivamente in la valutazione dei pazienti con citopatie mitocondriali who sottoporsi an experimental trattamento may non fornire an interamente accurate sense di efficacia della terapia. 31P risonanza magnetica spettroscopia può essere utilizzato per valutare energia parametri in the specifico tessuto of interest, che forniscono a noninvasive, quantitative misure of cervello e/o muscoli metabolismo. In the presenza of disordered metabolismo mitocondriale uno might expect per vedere a basso concentrazione of fosfocreatina, a alto concentrazione of inorganic fosfato, e a alto calcolato ADP.34 Muscle può essere esaminati a riposo, durante esercizio, e durante immediate postexercise recovery. In malattie of mitocondriale respirazione, a diminuire in the fosfocreatina/ inorganic fosfato (PCr/Pi) rapporto può essere osservata a riposo. C'è delayed replenishment of fosfocreatina a seguito esercizio. In alcune pazienti con citopatia mitocondriale ci può essere no anormalità on 31P MRS, probabilmente indicante che |
UMDF 24 Think Mitocondri
| muscoli scheletrici mitocondri non sono coinvolti.35 Again, con utilizzazione di queste tecnica, conflittuali rapporti exist come nel efficacia di CoQ10 terapia. Eight pazienti e 18 sano controls were trattati con 150 mg di CoQ10 al giorno per 6 mesi e esaminati by 31P MRS del calf muscoli a riposo, durante esercizio, e durante the postexercise recovery periodo. MRS was condotte a the beginning del trattamento e a seguito 3 e 6 mesi della terapia. The mean PCr/Pi was significantly più alte per controls prima to trattamento e did non significantly cambiano dappertutto the integrazione periodo. By 3 mesi del trattamento, there was a nonsignificant repletion of fosfocreatina nel popolazione di pazienti. Uno paziente had una drammatica miglioramento del PCr/Pi a riposo in aggiunta to aumentato repletion of fosfocreatina postexercise a seguito 3 mesi del trattamento.36 Barbiroli et al37 utilizzato in vivo phosphorous risonanza magnetica spettroscopia per valutare the efficacia di CoQ10 sul miglioramento cervello e muscoli scheletrici mitocondriale respirazione. Ten pazienti con citopatie mitocondriali were esaminati by 31P MRS prima to e 6 mesi a seguito trattamento con 150 mg al giorno di CoQ10. There were 36 età-matched, sano controls. Prior to trattamento tutti i pazienti dimostrato basso concentrazioni of fosfocreatina e alto ADP, indicative di disfunzione mitocondriale. There was a significante aumentato (P <0.02) cervello concentrazione of fosfocreatina a seguito trattamento con CoQ10, in aggiunta to a significante diminuire (P <0.01) of cervello concentrazione of inorganic phosphorous. Con regards nel muscoli scheletrici valutazione, the 31P MRS spectra were non significantly differenti a riposo either prima to o a seguito trattamento quando comparata con controls. Nonostante questo, tutti i pazienti did dimostrano a faster recovery of fosfocreatina a seguito trattamento e alcune (those con CPEO) riportati aumentato forza. Due pazienti con encefalomiopatia mitocondriale were trattati con 150 mg al giorno di CoQ10 e esaminati by bicycle ergometry e 31P nucleare risonanza magnetica (NMR) spettroscopia prima to e 10 mesi dopo initiation del trattamento. In entrambi pazienti prima trattamento there was a basso PCr/Pi a riposo, in aggiunta to a alto resting lattato serico. Acidosis occurred durante the esercizio fase, |
seguite da una ritardare in recovery dopo esercizio. Inoltre, the bicycle ergometer esame revealed a lowering del ventilatory soglia come pure riduzione del maximum ossigeno apporto. Pretreatment 31P NMR spettroscopia dimostrato the a seguito anormalità: twofold diminuire di ATP concentrazione e abnormally basso PCr/Pi rapporto. Following 10 mesi del trattamento, there was significante miglioramento durante the esercizio esame insieme con a diminuire in lattato a riposo, aumentare in consumo dell'ossigeno, aumentare in massimale load, e ventilatory soglia raggiunto normale gamma. 31P NMR spettroscopia (condotte on flexor digitorum superficialis muscoli) corroborated these rilevamenti in che there was a significante aumentare from the baseline PCr/Pi rapporto a riposo in aggiunta to migliorata recovery of tutti misurato parametri. The diminuita ATP concentrazione was still presenti, pensarono to a lesser grado. The postexercise PCr/Pi rapporto was essenzialmente unchanged per uno paziente ma dimostrato a quattrofold aumentare per the second paziente. These provoca lend supporto to l'efficacia of alto-dose administration di CoQ10.38 The letteratura suggerisce significante controversy riguardante l'efficacia di CoQ10 integrazione. Senza riguardo, molti pazienti riportano migliorata funzione, e the effetti collaterali associati con il suo uso sono rare. La maggioranza del trattamento clinici will somministrare a terapeutici sperimentazione in escalating dosi (4 to 15 mg per kg al giorno) per determinare la sua efficacia in una persona paziente. Idebenone Idebenone è an analog di CoQ10 e agisce entrambi come una radicale libero scavenger come pure stimolare ATP formazione by funzionamento come una mobile elettroni carrier. E' attualmente non disponibili in the United States. Un paziente con LHON e miopatia was trattati con orale idebenone (45 mg tre times al giorno con aumentare by 135 mg al giorno ogni 2 giorni) a seguito l'insorgenza of marcato spasticità e debolezza. By the sixth al giorno del trattamento (135 mg tre times al giorno), il paziente was in grado to walk, run, e climb stairs. On neurologici esaminazioni, there was marcato riduzione of spasticità del minore estremità in aggiunta to migliorata forza. Following osservazione di queste miglioramento, idebenone was continuati a a maximum of 405 mg al giorno per an ulteriori 3 mesi, durante con |
Think Mitocondri 25 UMDF
| il paziente remained clinicamente stabili. Following withdrawal of idebenone, il paziente di nuovo dimostrato debolezza del minore estremità e spastica paraparesis. Idebenone was reinitiated e entro 2 settimane miglioramento clinico was di nuovo osservata. Questo paziente was additionally esaminati by cervello e muscoli 31P MRS. Following approssimativamente 3 mesi del trattamento there was an aumentare in fosfocreatina concentrazione e a diminuire from baseline of inorganic fosfato toward reference valori. When reimaged a seguito withdrawal of idebenone, these parametri were marcatamente peggiorate e did non marcatamente migliora a seguito reinstitution of medicamento. In aggiunta, 3 settimane a seguito initiation del trattamento, muscoli studi mostrato an aumentato rate of recovery of fosfocreatina e inorganic fosfato. Though similare peggioramento of variables were osservata a seguito discontinuation di questo farmaco, once resumed there was no return nel previously osservata recovery livelli.39 A 10-anno-old ragazzo con LHON dovuta al omoplasmiche 11778 mutazione was trattati con orale idebenone (90 mg al giorno) dopo presentanti con precoce insorgenza sintomi. Dopo 7 mesi del trattamento, acuità visiva was migliorata slightly (6/90 bilaterally to 6/6).40 Comunque, spontaneous miglioramento in acuità visiva è frequentemente riportati in LHON. Un paziente con MELAS was trattati con CoQ10 augmented by the aggiunta of idebenone. Following 8 mesi del trattamento con 210 mg al giorno di CoQ10, il paziente dimostrato alcune miglioramento in termini of amelioration of sensoria disturbazione, atassia, e debolezza muscolare. Serum lattato diminuita slightly. Electroencephalogram (EEG) e Wechsler Adulti Intelligence Scale (WAIS) esaminazioni remained unchanged quando comparata to prima to trattamento. Motor e sensoria conduzione velocities normalizzati. Following the aggiunta of 90 mg of idebenone, muscoli forza migliorata ulteriori. Her EEG revealed marcato miglioramento from baseline e from durante trattamento con CoQ10 alone. Her WAIS scores aumentato by 14 punti. CSF proteine diminuita from 64 mg/dL to 45 mg/dL. The idebenone dose was poi aumentato to 180 mg al giorno per an ulteriori 11 mesi. il lattato nel CSF diminuita. The miglioramenti were mantenuti per a follow-up time of 20 mesi.41 The sperimentano con idebenone può essere troppo limitato |
to draw ogni definitive conclusioni. Ulteriori indagini may elucidate a ruolo per questo agente in a subset dei pazienti con una malattia mitocondriale. Levo-Carnitine Endogenous levo-carnitina (betahydroxy- gammatrimethylammonium butyrate), trovati in molti umana tessuti, è an amino acido derivative. E' sintetizzato in the fegato e reni from proteine-bound lisina (supplemental orale lisina cannot migliora carnitina sintesi) e methionine. E' a acqua-soluble sostanza che exhibits biologic attività solamente quando in the levo isoform. Numerose enzimi e cofattori (ferro, ascorbic acido, niacin, e pyridoxine) sono coinvolti in its biosintesi, e solamente uno matrice mitocondriale enzimi è coinvolti in the percorso. Of note, scheletrici e cuore muscoli sono incapace to synthesize carnitina, e these tessuti sono di conseguenza dipendente on apporto of carnitina from sangue. Normal plasma e tessuto livelli sono mantenuti by entrambi de novo sintesi e esogena dietetici fonti. Meat e latte prodotti contiene il più alto concentrazioni of dietetici carnitina laddove plant prodotti sono scarsa fonti. Normal plasma carnitina concentrazioni sono circa 25 umol/L in infanti e 54 umol/L in adulti.42 Le più alto concentrazione of carnitina è trovati in muscoli scheletrici (98%), sebbene distribuzione è shared con cuore, reni, fegato, e cervello.43 Carnitine è presenti in tessuti e fisiologico liquidi come either libera carnitina o come the acilcarnitina ester. In normale circostanze, approssimativamente 85 to 90% è presenti in the libera state. La maggioranza of plasma acilcarnitina è rappresentate by acetylcarnitine, con è spesso nonpathologically elevati in the digiuno state. Il rapporto fra acilcarnitina to libera carnitina varia con timing del last meal, composizione of che meal, nutrizionale status, esercizio, e malattia condizioni e è abbastanza sensibili to cambiamenti nel metabolismo mitocondriale. A rapporto of 0.25 è considerate di essere normale, laddove la più grande than 0.4 è anormale e è indicative of carnitina insufficienza o insufficienti carnitina alla luce del metaboliche domanda.44 Carnitine è necessarie per transporting a catena lunga grassi acidi across the membrane mitocondriali interne per il processo of beta-ossidazione. Questo avvengono principalmente in muscoli scheletrici, cuore, e fegato e è carried out by carnitina palmitoyltransferase I (CPT I), acilcarnitina traslocasi, e CPT II. A second |
UMDF 26 Think Mitocondri
| maggiore task of carnitina è a mantenere intracellulare omeostasi of acil-CoA. Carnitine transesterifies the acil-CoA esteri che è originato durante beta-ossidazione attraverso the action of carnitina-acyltransferases. The acilcarnitina can poi cross the membrane mitocondriali in scambio per libera carnitina, di conseguenza allowing per ristabilimento of libera CoA dentro i mitocondri. In aggiunta to these maggiore funzioni, carnitina may anche giocano alcune ruolo in alteranti the fisiologico proprietà delle membrane cellulari, tali come membrane stabilizzazione.45 In the setting of congeniti errori del metabolismo, carnitina serve to detoxify the poisonous metaboliche intermedi by forming a meno tossico ester. A numero of pathologic condizioni sono state associati con anormale metabolismo of carnitina, la più frequente of con è carnitina carenza. Carnitine carenza può essere definita come una state quando the concentrazione non è adeguate a raggiungere il corpo’s normale carnitina richiesta. Sistemico carnitina carenza può essere primaria ma può avvenire in molti malattia states, comprendenti malattie della fosforilazione ossidativa, betaoxidation, organiche acidurias, malnutrizione, valproato, e zidovudine use e in those receiving totale parenteral nutrizione senza adeguate carnitina rimpiazzamento. Molti metaboliche malattie portano a elevati livelli of acil-CoA intermedi, le quali danneggiano the funzione of adenine nucleotide traslocasi, the enzimi che scambios ADP per ATP across the membrane mitocondriali interne. Carnitine forme an ester linkage con the acil-CoA, forming the relativamente nontoxic acilcarnitina, con è escrete nelle urine. Elevati livelli of acil-CoA intermedi nel corso del tempo può portare a a secondari carnitina carenza. Likewise, a carnitina carenza stessa può provocare in aumentato tossicità del accumulate acil-CoA sostanze. Clinical manifestazioni of a carnitina-deficient state sono varied, comprendenti ma non limitato to cardiomiopatia, acute encefalopatia, miopatia, ritardo cognitivo, sistema nervoso centrale disfunzione, gastrointestinale dismotilità, e ricorrenti incidences of metaboliche decompensazione. Trattamento con levo-carnitina devono essere considerate per ogni persona con a primaria o secondari carnitina carenza. The ruolo of carnitina terapia in malattia mitocondriale è threefold. Come already discusse, carnitina plays a ruolo in reestablishing |
omeostasi of acil gruppi, a processo che è aberrante quando disfunzione mitocondriale exists, portante a inibizione of respiratoria enzimi. In aggiunta, secondari carnitina carenza exists in the setting of citopatie mitocondriali; di conseguenza, carnitina rimpiazzamento è l'essenziale. Infine, carnitina può fornire migliorata integrità del membrane mitocondriali, di conseguenza adding to membrane stabilizzazione.46 The tipico dose of levo-carnitina è 100 mg per kg al giorno per bambini e 2 to 4 grammi al giorno per adulti in tre divided dosi. In the nonacute setting, levo-carnitina è disponibili come una liquid o tablet, ma è anche disponibili come un intravenoso preparazione. The intravenoso dose è lo stesso come the orale dose. Prior to initiation of carnitina terapia in ogni paziente, plasma e urine carnitina e acilcarnitina profili devono essere ottenuto. The primaria effetti collaterali comprendono diarrea e nausea, sebbene carnitina è solitamente ben tollerato a tipico dosi. Dovrebbe essere notati che orale assorbimento è variabile, e come piccolo come 15% del orale dose may actually be assorbite. Of 48 pazienti studiati by Campos ed al.,47 quattro had entrambi totale e libera plasma carnitina carenza (entrambi definita come <30 μmol/L con normals in the basso to mid 50s) con carnitina insufficienza (definita come rapporto of esterificati to libera carnitina >0.25 con normale 0.13 ± 0.016), e 17 had isolato carnitina insufficienza. All 21 pazienti con carnitina carenza o insufficienza were trattati con 50 to 200 mg per kg al giorno of levocarnitina. The a seguito miglioramenti were osservata a seguito initiation del trattamento: 20 of 21 pazienti con debolezza muscolare dimostrato soggettiva miglioramento in muscoli tono, quattro of otto pazienti con crescita insufficiente mostrato crescita acceleration, e otto of otto pazienti con cardiomiopatia dimostrato migliorata echocardiographic rilevamenti e miglioramento clinico. La media trattamento durata was 11 mesi (gamma 1 to 24 mesi). Plasma livelli della carnitina 10 giorni dopo initiation del trattamento were normale o sopra normale. Molti ulteriori rapporti Hanno dimostrato gli effetti benefici of carnitina in the setting della cardiomiopatia dovuta al sottostanti metaboliche etiologies, comprendenti mitocondriale anormalità. La creatina La creatina è an amino acido prodotto endogenamente in the fegato from arginina e glicina, ed è anche trovati in meat prodotti. La creatina fosfato è synUMDF |
Think Mitocondri 27 UMDF
| thesized from creatina e ATP, ed è catalyzed by creatina chinasi (CK). Improbabili ATP, con il corpo è incapace to store, creatina fosfato può essere stored to a limitato grado in tessuti, allowing per a supply del alto-energia fosfato bond, le quali possono essere utilizzato quando necessaria. The idratazione of fosfocreatina to creatina e ATP thereby permette the ATP di essere utilizzato by il tessuto. La creatina è trovati in più alto concentrazioni in muscoli scheletrici e to lesser gradi in cardiaca muscoli, smooth muscoli, cervello, sperm, e reni. Intramuscular fosfocreatina può essere ridotti in pazienti con citopatie mitocondriali. Supplemental creatina sembri di essere più efficace a incrementare fosfocreatina e creatina in questo setting. Harris ed al. dimostrato che administration della creatina to sano soggetti resulted in a la più grande effetto per quei pazienti whose iniziale lal creatina totale concentrazione was basso. There were no effetti collaterali from integrazione con dosi ranging from 70 g to 330 g con maximum trattamento time of 21 giorni.48 The razionale per usando creatina è per aumentare il tessuto concentrazioni e possibilmente aumentare la capacità dei muscoli (o altro organi) to accumulate creatina fosfato. Tarnopolsky ed al. Hanno shown che creatina monoidrato (a dosi of 5 g twice al giorno per 2 settimane seguite by 2 g twice al giorno per 1 week) migliorata forza per alto-intensity anaerobico e aerobica attività e lean muscoli mass in pazienti con neuromuscolare malattie, comprendenti those con miopatia mitocondriale. Entrambi sperimentazioni were basato on breve termine provoca, e the longterm effetti benefici della creatina rimane di essere provato. Senza riguardo, l'uso della creatina in critiche situazioni sembri di essere ragionevole.49,50 In uno studio of nine sano men fornita con either orale creatina (come creatina monoidrato, 20 g dose) o placebo dimostrato no effetto on prestazione per maximum esercizio o on fosfocreatina livelli. Comunque, l'integratore was solamente somministrato over a 3-al giorno periodo e was in a setting of probabilmente normale muscoli creatina livelli; di conseguenza, its relevancy a quelle con una malattia mitocondriale non è conosciuta.51 Antioxidants Numerous pharmacologic agenti sono state usati nel trattamento of citopatie mitocondriali, comprendenti antiossidanti. Antioxidants può migliorare funzione enzimatica |
o lento il processo of ossidativa danno, sebbene the beneficio nel corso del tempo non è possibili per misurare. Le più comunemente employed antiossidanti (e the tipico al giorno dosaggi) per pazienti con una malattia mitocondriale comprendono selenio (50 to 100 mcg), vitamine C (250 to 4000 mg in divided dosaggi), vitamine E (400 to 1200 IU in divided dosaggi), e lipoic acido (200 to 600 mg in divided dosaggi). Dato the vast manifestazione clinica del citopatie mitocondriali e the fact che these agenti Hanno non systematically been studiati in questo setting, it non è possibili to state che ci sono provato beneficio. Nonostante questo, they sono routinariamente usati in pazienti con queste malattie. Riboflavin Riboflavin (vitamine B2) è a precursor to flavin mononucleotide (FMN) e flavin adenine dinucleotide (FAD), con sono cofattori of ETC complessi I e II, rispettivamente. Riboflavin è stata proposti to agire therapeutically by uno of numerosi potenziale meccanismi comprendenti inibizione del breakdown of complesso I by che forniscono più resistenza to proteolysis o stabilizing the membrane mitocondriali. E' stata usati con alcune success in alcune pazienti con una malattia mitocondriale senza ogni apparente effetti collaterali. A 33-anno-old paziente con MELAS e carenza del complesso I was somministrato a combinazione of riboflavina (100 mg tre times al giorno) e nicotinamide (1 g quattro times al giorno) in a doubleblinded, modo randomizzato. In conjunction con questo trattamento, there was cessation di queste paziente’s encephalopathic e miopatica sintomi. The spectroscopic rilevamenti were equivoci: resting PCr/Pi did non show ogni trattamento effetto, laddove alto-energia fosfato recovery deteriorated upon withdrawal of nicotinamide (ma non riboflavina). Sural nerve sensoria esaminazioni revealed a drop in ampiezza once terapia was withdrawn con recovery a seguito the reinitiation del trattamento (P = 0.00007 destra, 0.017 left).52 A 10-mese-old infante girl con a parziale difetto of complesso I was trattati con incrementare dosi of riboflavina (beginning a 3 mg per kg). It was osservata che livelli del lattato normalizzati e debolezza muscolare migliorata a the massimale dose of 13 mg per kg.53 Riboflavin was dati to five pazienti con miopatia mitocondriale dovuta al complesso I |
28 Think Mitocondri
| carenza in dosaggi ranging from 9 to 60 mg al giorno. i pazienti presentati con either pure miopatica sintomi o encefalomiopatia. Uno paziente had stabilizzazione del previously regressive malattia e tre mostrato miglioramento clinico, specialmente in the miopatica componente. There was normalization of l'attività del complesso I in tre del pazienti in aggiunta to migliorata livelli del lattato o muscoli histopathology.54 A 13-anno-old girl con progressive intolleranza all'esercizio, grave acidosi lattica, e carenza del complesso I was trattati con 100 mg of orale riboflavina al giorno. There was remarkable e persistente miglioramento clinico con an aumentare in tolleranza all'esercizio. Esercizio parametri (massimale lavoro capacità, O2 apporto, e base eccesso) anche migliorata.55 A 6-anno-old ragazzo con a difetto of complesso I e miopatia presentanti come lentamente progressive debolezza was con pieno successo trattati con riboflavina e carnitina. Clinically, muscoli forza e motor conduzione velocities were migliorata. In aggiunta, the l'attività del complesso I e livelli della carnitina normalizzati 7 mesi dopo the partenza del trattamento.56 Più recentemente, Ogle et al57 riportano on a 21⁄2-anno-old paziente con carenza del complesso I, DNA mitocondriale mutazione, e miopatia e who had persistente risposte to riboflavina terapia (20 to 25 mg twice al giorno) over a 3-anno periodo. Nonostante persistente acidosi lattica, questo paziente dimostrato miglioramenti in termini of overall muscoli forza e resistenza. Riboflavin non è sempre efficace, come dimostrato by il caso of a 4-mese-old infante con severecongenital acidosi lattica e carenza del complesso I, who underwent una sperimentazione of riboflavina a a dose of 100 mg al giorno. Il bambino’s lattato serico e rapporto a piruvato remained significantly elevati, e il paziente continuati to dimostrano caratteristiche of grave miopatia e cardiomiopatia fino a che his morte.58 quattro pazienti con MELAS in associazione con carenza del complesso I trattati con riboflavina, altro integratori, e the dieta chetogena did non show miglioramento. 59 Un altro grande sperimentazione of riboflavina in combinazione con altro integratori trovati no significante terapeutici success.33 The provoca del trattamento con riboflavina, con a grande variazioni in dosi e trattamento durata in questo diverse popolazione, non sono uniformi ma dimostrano che those con carenza del complesso I e miopatia pura possono beneficiare from supplemental riboflavina, con o senza altro integratori. Naturalmente, the decorso clinico di queste |
malattie rimane variabile tali che ogni miglioramento osservata può non essere dovuta al interventi terapeutici. Tiamina The PDH complesso catalyzes the thiaminedependent decarboxylation of piruvato. Tiamina pyrophosphate, the physiologically attive forma di tiamina, agisce come una coenzima per questo decarboxylation. L'adozione di tiamina è stata stabiliti nel trattamento of alcune forme of PDH carenza, sebbene il suo uso in malattie coinvolgenti più distal componenti del metabolismo energetico, tali come catena di trasporto degli elettroni malattie, non è stabiliti.Non ci sono riportati effetti collaterali con administration. Following trattamento over a numerosi week periodo con tiamina a 300 mg tre times al giorno, tre pazienti con sindrome di Kearns-Sayre were trovati to Hanno normalization of previously anormale i livelli di lattato e piruvato. There was alcune cambiano in overall livello of affaticabilità, ma miglioramento clinico in generale was trivial.60 Un altro paziente, a 23-anno-old con miopatia mitocondriale, cardiomiopatia, e acidosi lattica, was trattati con tiamina (100 mg due times al giorno) in combinazione con prednisone (60 mg al giorno). Over a 3-week periodo, il paziente dimostrato progressive miglioramento in overall forza. The previously trattamento a vita episodi of acidosi lattica anche ceased, a rilevamenti che persisted over a 7-anno follow-up periodo.61 Uno larger studio dei pazienti con citopatie mitocondriali failed to dimostrano efficacia del trattamento a dosi of 100 mg al giorno per 2 mesi.33 Vitamina K3 In vitro studi show che in presenza of complesso I inibitori, vitamine K3 (menadione) stimulates ossigeno utilizzazione in mitocondri, derivante in an aumentare of NADH ossidazione.62 Uno gruppo of investigatori utilizzato 31P nucleare risonanza magnetica per valutare the risposte to trattamento con menadione (40 to 80 mg al giorno) e ascorbato (4 g al giorno) of a 19-anno-old paziente con miopatia mitocondriale associati con complesso III carenza. Questo trattamento approccio resulted in entrambi clinica e metaboliche miglioramento che persisted a 1-anno follow-up. She was no più a lungo wheelchairbound sebbene overall her debolezza remained unchanged. The clinica miglioramenti were sup |
Think Mitocondri 29 UMDF
| ported by 31P NMR dati, con revealed an aumentare in fosfocreatina concentrazione a riposo e postexercise. A doubling del iniziale dose of K3 migliorata the phosphorous NMR spectra persino ulteriori senza apparente effetti collaterali. Sintomi deteriorated con withdrawal del trattamento con recovery of funzione once reinstated.63,64 Toscano et al65 trattati a 16-anno-old girl con atassia, miocloni, acidosi lattica, e complesso III carenza con vitamine K3 (40 mg al giorno) e C (4 g al giorno). There was no significante cambiano in acidosi lattica ma there was lievi miglioramento dal suo atassia. Dopo 5 mesi della terapia, il cervello 31P-MRS indices were restored to normale. Skeletal muscoli valutazione, sebbene, revealed solamente slight miglioramento of mitocondriale bioenergetics. Uno larger sperimentazione nelle quali pazienti were trattati con 60 mg al giorno of menadione e 2 g al giorno di vitamine C (in aggiunta to multiple altro integratori) did non substantiate the terapeutici beneficio visto by these altro investigatori.33 K3 è conosciuta to cause hemolytic anemia, hyperbilirubinemia, e kernicterus e è di conseguenza controidicate per neonates, pregnant females, o those being trattati con coumadin. E' anche difficile to trovano, e K1, the comune forma di vitamine K, may Hanno no beneficio. Come a pratici alternative, CoQ10 può fornire lo stesso beneficio. Folate A 23-anno-old woman con sindrome di Kearns-Sayre on phenytoin per a dall'attacco epilettico disturbo was trovati to Hanno diminuita CSF e plasma folate livelli in aggiunta to basso libera carnitina. She was trattati con folate (40 μg per kg of corpo peso o 15 mg al giorno), D, levocarnitina (10 g al giorno), e methionine (500 mg al giorno). Plasma folate aumentato to normale con integrazione ma CSF folate remained unchanged. She anche dimostrato miglioramento clinico nel point of being in grado to ambulate dopo avendo been bedridden.66 Uridine Uridine will appena be sotto investigazione come una trattamento per malattie mitocondriali. Uridine è a pirimidina nucleotide, necessari per sintesi of RNA e DNA. Normal cellula e organo funzione rely on adeguate sintesi, trasporto, e interconversions of pyrimidines. The sintetica percorso per |
uridina sintesi implica the mitocondriale deidrogenazione of dihydrooroate to oroate, con è intimately collegata con CoQ10 recycling e normale catena di trasporto degli elettroni funzione. Ogni processo che interferes con CoQ10 recycling o catena di trasporto degli elettroni funzione can impair oroate formazione. Il processo di uridina sintesi concludes con condensing orotic acido con phosphoribosyl pyrophosphate (PRPP) to forma uridina monophosphate. Disordered fosforilazione ossidativa will impede the de novo sintesi of pyrimidines e ulteriori esacerbare cellulari disfunzione. The teorici argomento per trattamento con esogena uridina è to overcome la relativa deficit dovuta al danneggiato sintesi portante a migliorata cellulari health. ereditaria orotic aciduria provoca a primaria uridina carenza e provoca in a sindrome of crescita insufficiente, anemia megaloblastica, orotic aciduria, congenital malformations, transitori immunoglobulina carenza, immune carenza, e ritardo nello sviluppo. Lifelong supplemental uridina è necessari e may reverse alcune del manifestazioni di queste deficient state. Postulated effetti benefici di uridina sono basato primariamente on animal ricerche e comprendono: appetite stimolazione, anticonvulsivi effetti, prevenzione of acido lattico overproduction, antidepressant, cardioprotective, e prevenzione of cerebrale edema, fra altri. Human sperimentazioni sono planned. Side effetti sono state riportati e più includono solitamente a transitori aumentare in dall'attacco epilettico attività upon initiation della terapia, nausea, vomito, e/o diarrea (Robert Naviaux, personal comunicazione, 2001). Combination Therapy/Miscellaneous 1. Sixteen pazienti con miopatia mitocondriale, sindrome di Kearns-Sayre , MELAS, o MERRF were trattati con multiple integratori comprendenti vitamine K3 (20 to 60 mg al giorno), vitamine C (1 g due volte al giorno), α-tocoferolo (200 IU due volte al giorno), CoQ10 (30 to 120 mg al giorno), e methylprednisolone (2 to 16 mg ogni altro al giorno). All pazienti underwent a baseline 31P-NMR spettroscopia, sebbene solamente five were esaminati durante trattamento con questo metodo. Ten pazienti were esaminati usando near-infrarosso spettroscopia. Nessuna delle due the 31P-NMR spettroscopia nor near-infrarosso spettroscopia |
UMDF
30 Think Mitocondri
| dimostrato ogni acute cambiamenti in associazione con terapia. Follow-up gamma was 0.5 mesi to 15 anni. Ten del pazienti died. Nonostante the mancanza di obiettiva dati to dimostrano efficacia con questo interventi, the autori felt there was a subset dei pazienti who appeared to beneficio from questo trattamento con prolungato survival, meno funzionale disability, e/o fewer medica complicazioni.67 2. Thirteen pazienti trattati con al giorno dosi of levo-carnitina (100 mg to 200 mg), ubichinone (80 to 300 mg), tocoferolo (50 to 100 mg per kg), riboflavina (50 to 100 mg), vitamine K3 (80 to 160 mg), e vitamine C (2 g) underwent monitorizzazione of siero carnitina, ubichinone, e erythrocyte tocoferolo livelli. Total e livelli della carnitina libera aumentato con trattamento (P <0.05, totale carnitina) ma the acilcarnitina/libera carnitina indice dimostrato sustained innalzamento sopra che of controls (implying an aumentare of acilcarnitina generation). Serum ubichinone e erythrocyte tocoferolo livelli were più alte dopo trattamento, sebbene questo did non reach valore statistico. Additionally, livelli del lattato nel sangue were significantly diminuita in quattro pazienti who clinicamente either stabilized o migliorata (P <0.05).68 3. Lipoic acido funzioni come una coenzima del piruvato deidrogenasi complesso. In uno studio, trattamento of a 33-anno-old woman con CPEO con 600 mg al giorno of lipoic acido per 7 mesi was associati con a soggettiva miglioramento in sintomi e obiettiva cambiamenti in 31P NMR spettroscopia of cervello e muscoli. At baseline questo paziente dimostrato ridotti fosfocreatina concentrazione in conjunction con aumentato ADP concentrazione on cervello MRS. Following trattamento con lipoic acido there was an aumentato fosfocreatina contenuto e diminuita ADP, indicante che il cervello was funzionamento sotto più stabili condizioni.Additionally, alcune miglioramenti sono stati visti con muscoli 31P NMR spettroscopia sebbene non come impressive (rate of fosfocreatina resynthesis postexercise did non migliora dopo initiation della terapia).69 Summar Evaluating l'efficacia of trattamenti per citopatie mitocondriali è influenzato by la relativa rarità di queste condizioni e loro variabile e imprevedibile naturale course, come pure loro |
ampia clinica e biochimica eterogeneità. The mancanza di conclusive evidenze in favor di ogni uno trattamento, o combinazioni of trattamenti, rendono it impossibile per determinare conclusively con metaboliche terapie might be efficace. E' importante to note che non c'è evidenze suggesting terapia alters the ultimate course di queste altrimenti potenzialmente progressive malattie. C'è evidenze to suggerire alcune pazienti may Hanno an miglioramento in sintomi e an migliorata qualità della vita. La maggioranza di dati disponibili con regards to terapie specifice è anedotticamente, sebbene più recentemente c'è stata an emphasis on sperimentazioni cliniche controllate. Sfortunatamente, persino in questo setting, il popolazione di pazienti varia significantly e statistico constraints make it impossibile per valutare tutti potenziale risposte. Siccome malattie mitocondriali Hanno così molti potenziale sintomi, identifying con sintomo o sign per valutare come parte di ogni studio è problematici. Se uno studio relies on uno endpoint, miglioramenti in altro funzioni può essere ignored e miss a potenziale beneficio of a trattamento. Tentando per misurare ogni sintomo e sign del malattia sarebbe così burdensome che uno studio sarebbe prohibitively costosi. Come un esempio, relying on siero o CSF misurazioni del lattato alone may non pienamente dimostrano il completo terapeutici beneficio di una particolare medicamento. It ha anche been dimostrato in molti del studi discusse che persino con diminishment del livelli del lattato, there non è necessariamente concurrent sintomatico miglioramento. Pertanto, l'uso of altro obiettiva misure e possibilmente molti obiettiva misurazioni (cioè, nucleare risonanza magnetica spettroscopia of cervello e/o muscoli e bicycle ergometry) per valutare l'efficacia di queste interventi rimane cruciale to proving o disproving loro beneficio. Determination of clinica miglioramenti should anche be inclusi in an sforzo to prove a collegamento fra the biochimica e funzionale miglioramenti. Nonostante the mancanza di coerente dati, che forniscono integratori come parte di una persona sperimentazione nelle quali il paziente serve come loro own control sembri di essere a ragionevole approccio. The clinician e paziente will necessitano da usare loro meglio judgment come nel problemi di efficacia e costo. L'adozione dei farmaci tali come dicloroacetato è still sotto investigazione e will probabilmente rimane riservata per quei pazienti con |
Think Mitocondri 31 UMDF
| trattamento a vita acidosi lattica che non è responsive to convenzionale trattamento. Uridine rimane sotto investigazione con regards to its utilità nel trattamento of citopatie mitocondriali. |
UMDF 32 Think Mitocondri
Riferimenti
1. Chinnery PF, Howell N, Lightowlers RN, Turnbull
DM. Molecular pathology della MELAS e
MERRF: the relationship fra mutazione load
e clinica fenotipi. Cervello 1997;
120:1713–1721
2. Lauwers MH, Van Lersberghe C, Camu F.
Inhalation anaesthesia e the sindrome di Kearns-Sayre .
Anaesthesia 1994;49:876–878
3. Burns AM, Shelly MP. Anaesthesia per pazienti
con miopatia mitocondriale. Anaesthesia
1989;44:975–977
4. D’Ambra MN, Dedrick D, Savarese JJ. sindrome di Kearns- Sayre e pancuronium-succinilcolina-
indotta blocco neuromuscolare.
Anesthesiology 1979;51:343–345
5. Casta A, Quackenbush EJ, Houck CS, ed al.
Perioperative materia bianca degenerazione
e morte
in un paziente con a difetto in fosforilazione ossidativa mitocondriale. Anesthesiology
1997;87:420–425
6. Kitoh T, Mizuno K, Otagiri T, Ichinose A, Sasao J,
Goto H. Anesthetic gestione per un paziente
con sindrome di Kearns-Sayre . Anesth Analg
1995;80:1240–1242
7. Barohn RJ, Clanton T, Sahenk Z, Mendell JR.
Recurrent insufficienza respiratoria e depressed
ventilatory drive complicating miopatie mitocondriali. Neurology 1990;40:103–106
8. Maslow A, Lisbon A. Anesthetic considerazioni in
pazienti con disfunzione mitocondriale. Anesth
Analg 1993;76:884–886
9. Schenkman KA, Yan S. Propofol compromissione of
mitocondriale respirazione in isolato perfused
guinea pig hearts determinata by reflectant spettroscopia.
Crit Care Med 2000;28:172–177
10. Przyrembel H. Therapy delle malattie mitocondriali.
J InheritMetab Dis 1987;10(suppl 1):129–146
11. Taivassalo T, De Stefano N, Argov Z, ed al.
Effects of aerobica training in pazienti con
miopatie mitocondriali. Neurology
1998;50:1055–1060
12. Stacpoole PW, Henderson GN, Yan Z, Cornett
R, James MO. Pharmacokinetics, metabolismo,
e toxicology of dicloroacetato. Farmaco Metab
Rev 1998;30:499–539
13. Stacpoole PW, Barnes CL, Hurbanis MD,
Cannon SL, Kerr DS. Trattamento of congenital
acidosi lattica con dicloroacetato. Arch Dis
Child 1997;77:535–541
14. Stacpoole PW, Lorenz AC, Thomas RG,
Harman EM. Dichloroacetate nel trattamento
of acidosi lattica. Ann Inter Med
1988;108:58–63
15. Stacpoole PW, Wright EC, Baumgartner TG, ed al. A sperimentazione controllata clinica of dicloroacetato
per trattamento of acidosi lattica in adulti. N Engl
J Med 1992;327:1564–1569
16. Kuroda Y, Ito M, Naito E, ed al. Concomitant
administration di sodio bicarbonato dicloroacetato e
vitamine B1 per lattica acidemia in bambini con
MELAS sindrome. J Pediatr
1997;131:450–452
17. Saijo T, Naito E, Ito M, Takeda E, Hashimoto
T, Kuroda Y. Therapeutic effetto di sodio bicarbonato
dicloroacetato on visual e auditory allucinazioni
in un paziente con MELAS.
Neuropediatrica 1991;22:166–167
18. Kimura S, Osaka H, Saitou K, Ohtuki N,
Kobayashi,Nezu A. Il miglioramento di lesioni
shown on MRI e CT scansione by administration
of dicloroacetato in pazienti con sindrome di Leigh.
J Neurol Sci 1995;134:103–107
19. Tulinius MH, Eriksson BO, Hjalmarson O,
Holme E, Oldfors A. Mitocondriale miopatia
e cardiomiopatia in fratelli e sorelle. Pediatr Neurol
1989;5:182–188
20. Takanashi J, Sugita K, Tanabe Y, Maemoto T,
Niimi H. Dichloroacetate trattamento in sindrome di Leigh causato by DNA mitocondriale mutazione.
J Neurol Sci 1997;145:83–86
21. DeStefano N, Matthews PM, Ford B, Genge A,
Think Mitocondri 33 UMDF
Karpati G, Arnold DL. Short-termine dicloroacetato
trattamento migliora indices of cerebrale
metabolismo in pazienti con malattie mitocondriali.
Neurology 1995;45:1193–1198
22. Pavlakis SG, Kingsley PB, Kaplan GP,
Stacpoole PW, O’Shea M, Lustbader D.
risonanza magnetica spettroscopia: use in monitorizzazione
MELAS trattamento. Arch Neurol
1998;55:849–852
23. Kimura S, Ohtuki N, Nezu A, Tanaka M,
Takeshita S. Clinical e radiologic miglioramenti
in encefalomiopatia mitocondriale a seguito
sodio bicarbonato dicloroacetato terapia. Cervello
Dev 1997;19:535–540
24. Ogasahara S, Engel A, Frens D, Mack D.
Muscle coenzima Q carenza in familial
encefalomiopatia mitocondriale . Proc Natl
Acad Sci U S A 1989;86:2379–2382
25. Goda S, Hamada T, Ishimoto S, Kobayashi T,
Goto I, Kuroiwa Y. Clinical miglioramento dopo
administration di coenzima Q10 in un paziente
con encefalomiopatia mitocondriale . J
Neurol 1987;234:62–63
26. Ogasahara S, Yorifuji S, Nishikawa Y, ed al.
Il miglioramento of anormale metabolismo del piruvato
e cardiaca difetto di conduzione con coenzima
Q10 in sindrome di Kearns-Sayre . Neurology
1985;35:372–377
27. Bresolin N, Bet L, Binda A, ed al. Clinical e
biochimica correlations in miopatie mitocondriali trattati con coenzima Q10.
Neurology 1988;38:892–899
28. Abe K, Fujimura H, Nishikawa Y, ed al. Marked
riduzione nel lattato nel CSF e livelli del piruvato
dopo CoQ terapia in un paziente con miopatia mitocondriale, encefalopatia, acidosi lattica
e episodi di simil-ictus (MELAS). Acta
Neurol Scand 1991;83:356–359
29. Chen RS, Huang CC, Chu NS.
Coenzima Q10
trattamento in mitocondriale encephalomyopathies:
breve termine doubleblind, crossover
studio. Eur Neurol 1997;37:212–218
30. Chan A, Reichmann H, Koegel A, Beck A,
Gold R. Metaboliche cambiamenti in pazienti con
miopatie mitocondriali e effetti di coenzima
Q10 terapia. J Neurol 1998;245:681–685
31. Bresolin N, Doriguzzi C, Ponzetto C, ed al.
Ubidecarenone nel trattamento delle miopatie mitocondriali: a multi-centri a doppio cieco sperimentazione.
J Neurol Sci 1990;100:70–78
32. Zierz S, von Wersebe O, Bleistein J, Jerusalem
F. Exogenous coenzima Q (CoQ) fails per aumentare CoQ in muscoli scheletrici di due pazienti
con miopatie mitocondriali. J Neurol Sci
1990;95:283–290
33. Matthews PM, Ford B, Dandurand RJ, ed al.
Coenzima Q10 con multiple vitamine è generalmente
inefficace in trattamento di malattia mitocondriale. Neurology 1993;43:884–890
34. Eleff SM, Barker PB, Blackband SJ, ed al.
Phosphorous risonanza magnetica spettroscopia
dei pazienti con citopatie mitocondriali
dimostra diminuita livelli of cervello fosfocreatina.
Ann Neurol 1990;27:626–630
35. Argov Z, Bank WJ. Phosphorous risonanza magnetica
spettroscopia (31P MRS) in neuromuscolare
malattie. Ann Neurol 1991;30:90–92
36. Gold R, Seibel P, Reinelt G, ed al. Phosphorous
risonanza magnetica spettroscopia in la valutazione
delle miopatie mitocondriali: provoca of a
6-mese terapia studio con coenzima Q. Eur
Neurol 1996;36:191–196
37. Barbiroli B, Iotti S, Lodi R. Improved cervello e
muscoli mitocondriale respirazione con CoQ: an
in vivo studio by 31P-MR spettroscopia in
pazienti con citopatie mitocondriali. Bio-
I fattori 1999;9:253–260
38. Bendahan D, Desnuelle C, Vanuxen D, ed al. 31P
NMR spettroscopia e ergometer esercizio esame
come evidenze per muscoli ossidativa prestazione
miglioramento con coenzima Q in miopatie mitocondriali. Neurology 1992;42:1203–1209
39. Cortelli P, Montagna P, Pierangeli G, ed al.
Clinical e cervello bioenergetics miglioramento
con idebenone in un paziente con neuropatia ottica ereditaria di Leber : a clinica e 31P-MRS
studio. J Neurol Sci 1997;148:25–31
40. Mashima Y, Hiida Y, Oguchi Y. Remission of
UMDF 34 Think Mitocondri
neuropatia ottica ereditaria di Leber con
idebenone. Lancet 1992;340:368–369
41. Ihara Y, Namba R, Kuroda S, Sato T, Shirabe T.
Mitocondriale encefalomiopatia (MELAS):
patologiche studio e successo terapia con
coenzima Q10 e idebenone. J Neurol Sci
1989;90:263–271
42. Schmidt-Sommerfield E, Werner D, Penn D.
Carnitine plasma concentrazioni in 353 metabolicamente
sano bambini. Eur J Pediatr
1988;147:356–360
43. Angelini C, Vergani L, Martinuzzi A. Clinical
e biochimica aspetti of carnitina carenza
e insufficienza: trasporto difetti e congeniti
errori of beta-ossidazione. Crit Rev Clin Lab Sci
1992;29:217–242
44. Pons R, DeVivo DC. La principale e secondari
carnitina carenza sindromi. J Child
Neurology 1995;10(suppl 2):9–24
45. Carter AL, Abney TO, Lapp DF. Biosynthesis
e metabolismo of carnitina. J Child Neurol
1995;10(suppl 2):3–7
46. Bellei M, Battelli D, Guarriero DM, ed al.
I cambiamenti in mitocondriale attività causato by
ammonium salts e the protective effetto of
carnitina. Biochem Biophys Res Commun
1989; 158:181–188
47. Campos Y, Huertas R, Lorenzo G, ed al. Plasma
carnitina insufficienza e efficacia of Lcarnitine
terapia in pazienti con miopatia mitocondriale. Muscle Nerve 1993;16:150–153
48. Harris RC, Soderlund K, Hultman E. Elevation
della creatina in resting e esercitata muscoli of
normale soggetti by dall'integrazione con creatina.
Clin Sci 1992;83:367–374
49. Tarnopolsky MA, Roy BD, MacDonald JR. A
randomizzati, sperimentazione controllata della creatina monoidrato
in pazienti con citopatie mitocondriali. Muscle Nerve 1997;20:1502–1509
50. Tarnopolsky MA, Martin J. La creatina monoidrato
aumento forza in pazienti con neuromuscolare
malattia. Neurology 1999;52:854–857
51. Odland LM, MacDougall JD,Tarnopolsky MA,
Elorriaga A, Borgmann A. The effetto of orale Cr
integrazione sui muscoli [PCr] e shortterm
massimale power output. Med Sci Sports
Exerc 1997;29:216–219
52. Penn AMW, Lee JWK, Thuillier P, ed al.
MELAS sindrome con mitocondriale
tRNALeu(UUR) mutazione: correlazione of clinica
state, conduzione nervosa, e muscoli 31P
risonanza magnetica spettroscopia durante trattamento
con nicotinamide e riboflavina.
Neurology 1992;42:2147–2152
53. Griebel V, Kraegeloh-Mann I, Ruitenbeek W,
Trijbels JMF, Paulus W. A miopatia mitocondriale in an infante con acidosi lattica. Dev
Med Child Neurol 1990;32:528–531
54. Bernsen PLJA, Gabreels FJM, Ruitenbeek W,
Hamburger HL. Trattamento di carenza del complesso I
con riboflavina. J Neurol Sci
1993;118:181–187
55. Arts WFM, Scholte HR, Bogaard JM, Kerrebijn
KF, Luyt-Houwen IEM. NADH-CoQ riduttasi
deficient miopatia: successo trattamento con
riboflavina. Lancet 1983;2:581–582
56. Bernsen PLJA, Gabreels FJM, Ruitenbeek W,
Sengers RCA, Stadhouders AM, Renier WO.
Successful trattamento of miopatia pura, associati
con carenza del complesso I, con riboflavina
e carnitina. Arch Neurol 1991;48:334–338
57. Ogle RF, Christodoulou J, Fagan E, ed al.
Mitocondriale miopatia con tRNALeu(URR)
mutazione e carenza del complesso I responsive
to riboflavina. J Pediatr 1997;130:138–145
58. Hoppel CL, Kerr DS, Dahms B, Roessmann U.
Deficiency del ridotti nicotinamide adenine
dinucleotide deidrogenasi componente of complesso
I of trasporto mitocondriale degli elettroni. J
Clin Invest 1987;80:71–77
59. Ichiki T, Tanaka M, Nishikimi M, ed al.
Deficiency of subunità of complesso I e encefalomiopatia mitocondriale . Ann Neurol
1988;23:287–294
60. Lou HC. Correction of aumentato plasma piruvato
e lattato plasmatico livelli usando grande dosi
di tiamina in pazienti con sindrome di Kearns-Sayre .
Arch Neurol 1981;38:469
61. Mastaglia FL, Thompson PL, Papadimitriou JM.
Mitocondriale miopatia con cardiomiopatia,
acidosi lattica: risposte to prednisone e
tiamina. Aust N Z J Med 1980;10:660–664
Think Mitocondri 35 UMDF
62. Cooper JM, Hayes DJ, Challiss RAJ, Morgan-
Hughes JA, Clark JB. Trattamento of experimental
NADH ubichinone riduttasi carenza con
menadione. Cervello 1992;115:991–1000
63. Eleff S, Kennaway NG, Buist NRM, ed al. 31P
NMR studio of miglioramento in fosforilazione ossidativa
by vitamine K3 e C in un paziente
con a difetto in trasporto degli elettroni a complesso
III in muscoli scheletrici. Proc Natl Acad Sci U S
A 1984;81:3529–3533
64. Argov Z, Bank WJ, Maris J, ed al..Trattamento of
miopatia mitocondriale dovuta al complesso III
carenza con vitamine K3 e C: a 31PNMR
follow-up studio. Ann Neurol
1986;19:598–602
65. Toscano A, Fazio MC, Vita G, ed al. Early-insorgenza
atassia cerebellare, miocloni e ipogonadismo
in a caso of mitocondriale complesso III carenza
trattati con vitamine K3 e C. J Neurol
1995;242:203–209
66. Allen RJ, DiMauro S, Coulter DC,
Papadimitriou A, Rothenberg SP. sindrome di Kearns-Sayre con ridotti plasma e cerebrospinal
liquidi folate. Ann Neurol
1983;13:679–682
67. Peterson PL. Il trattamento delle miopatie mitocondriali e encephalomyopathies.
Biochimica Biophysica Acta
1995;1271:275–280
68. Artuch R, Vilaseca MA, Pineda M. Biochemical
monitorizzazione del trattamento in paediatric
pazienti con una malattia mitocondriale. J Inherit
Metab Dis 1998;21:837–845
69. Barbiroli B, Medori R, Tritschler HJ, ed al.
Lipoic (thioctic) acido aumento cervello energia
disponibilità e muscoli scheletrici prestazione come
shown by in vivo 31P-MRS in un paziente con
citopatia mitocondriale. J Neurol
1995;242:472–477
Adulti Presentations of Mitocondriale malattia
by Robert K. Naviaux, M.D., Ph.D.
The Mitocondriale e Metaboliche malattia Center
University of California, San Diego
© The Mitocondriale News, United Malattie mitocondriali Foundation, 2000
| Mitocondriale medicine ha diventata uno del fastest growing nuovi disciplines in medicine (Luft 1994; 1995, Graff 1999). New malattie mitocondriali sono being descritte ogni anno. Nearly uno hundred differenti mutazioni in DNA mitocondriale sono state descritte, e nearly 500 nucleare gene difetti sono associati con disfunzione mitocondriale. Non tutti di queste genetica difetti cause measurable declines in fosforilazione ossidativa – il processo by con cibo e ossigeno sono combinate a fare energia (ATP). Tuttavia, persino the malattie mitocondriali che do non cause measurable energia failures può essere catastrofica e difficile per diagnosticare. Our expanding knowledge del molecolare, biochimica, e caratteristiche cliniche delle malattie mitocondriali ha forzati a cambiano in how scienziati understand these complesso malattie. Questo articolo will rassegna alcune del hallmarks di queste malattie in adulti, e outline alcune del esami che sono necessari per diagnosi. “Ogni malattia. Ogni organo. Ogni età.” Questo è forse the meglio generale summary dello spettro di malattia mitocondriale disponibili (Christodoulou 1999). Mitocondriale malattie sono notorious masqueraders (Kerr 1998). They può causare sintomi che sono indistinguishable from those causato by comune malattie. Solo the comportamento del malattia mitocondriale nel corso del tempo sets it apart from its più comune cousins. Mitocondriale disfunzione ha ora been collegata to comune maladies come diverse come infertility (Jansen 1998), cancro (Susin 1998), emicranie cefalee (Welch 1995), diabete (Damore 1999), malattia cardiaca (Hatch 1998, DiMauro 1998), cecità (Latkany 1999), sordità (Fischel 1999), reni malattia (Niaudet 1996), malattia epatica (Treem 1998), ictus (Heales 1999), the tossicità of AIDS farmaci (Barile 1998), Parkinson malattia (Kosel 1999), Alzheimer demenza (Fiskum 1999), e the invecchiamento processo stessa (Wallace 1997). Gli studi epidemiologici Hanno stabiliti al di fuori doubt che quando these cronica malattie sono studiati come gruppi, they |
sono complesso e Hanno molteplici cause – entrambi genetica e ambientale. La malattia mitocondriale non deve cause a majority di ogni uno del malattie elencato. Comunque, è importante ricordare che malattia mitocondriale può essere a cause di ogni di queste malattie, perché malattia mitocondriale tends to extend to altro sistemi d'organo, procedere con età, e rispondono poorly to current terapia. A comprehensive listing of segni e sintomi di malattia mitocondriale è al di fuori dello scopo di questo articolo. Questo è vera in parte perché the generale statement quoted a the beginning di queste paragraph è a clinica fact. Le più pazienti we valutarne con provato malattia mitocondriale, the broader the spettro of segni e sintomi diventa che we normalmente associate con ogni particolare malattia. Alcuni esempi sarà reviewed sotto to clarify questo point. Persino una singola point mutazione in DNA mitocondriale può produrre molti differenti malattie. Perhaps the meglio studiati esempio di questo è la mutazione A3243G first collegata nel malattia chiamati MELAS (encefalomiopatia mitocondriale , lattica acidemia, episodi di simil-ictus ). In our sperimentano a the Mitocondriale e Metaboliche malattia Center, adulti bearing questo mutazione in DNA mitocondriale frequentemente presentati con diabete anni prima insorgenza of cervello malattia. Alcuni pazienti first sofferto malattia psichiatrica e perdita di udito per decadi prima l'insorgenza of ricorrenti episodi di simil-ictus e diabete led nel correct diagnosi. Altro pazienti had precoce insorgenza demenza in loro 30s che was non diagnosticata fino a che the occurrence of a dall'attacco epilettico e simil-ictus episodio. In still altro pazienti, the singola manifestazione di queste mutazione was an inspiegata cardiomiopatia e mildly elevati lattato. Nearly a third del pazienti who carry la mutazione A3243G had near normale acido lattico serico livelli. In questi pazienti, solamente the cerebrospinal liquidi acido lattico was elevati. In 10-15%, acido lattico was elevati neither nel sangue nor spinal liquidi. The message è che non tutti i pazienti |
UMDF 36 Think Mitocondri
| con la mutazione A3243G Hanno MELAS. Similarmente, la maggior parte delle adulto pazienti who carry the T8993G mutazione, spesso riferite to come the NARP mutazione, do non Hanno the “neuropatia o neurogenic muscolare debolezza, atassia, o retinite pigmentosa”, per i quali the acronym was coined. These facts illustrate che the name di una malattia mitocondriale può essere ingannevole. Medici e pazienti who rely on an acronym like MELAS o NARP to guide them in making a diagnosi sarà wrong più spesso than non. Persino classica malattie mitocondriali like sindrome di Kearns-Sayre può essere difficile per diagnosticare quando la prima sintomi appare. Nessuna delle due l'insorgenza nor the rate of progressione to altro sistemi d'organo è stereotyped. When ptosi, oftalmoplegia, retinopatia, atassia, debolezza, affaticabilità all'esercizio, cardiaca conduzione blocco, elevati cerebrospinal liquidi proteine, e sfilacciate rossi fibre sono tutti presenti insieme, la diagnosi è semplice. Comunque, it may take decadi of lento progressione prima tutti these sintomi sono presenti insieme. Più spesso in adulti, i sintomi appariranno uno by uno over numerosi anni. When progressione è relativamente lento, adulto pazienti will tipicamente be riferite to un nuovo medica specialista ogni few anni, to take care di ogni nuovi sintomo come it appears. Spesso è a medica student cercando a fare sense del multisistemiche malattia who prompts a rivolgendosi a mitocondriale e malattia metabolica centri quando the final diagnosi è composta. Se malattie mitocondriali sono così complesso e proteiformi, how can generale medici make la diagnosi? A sistematica approccio è l'essenziale. It sarebbe wrong to say che everyone con diabete o malattia cardiaca devono essere checked per la malattia mitocondriale. Tabella 1 elenca tre rules of thumb che possono aiutare guide anyone who suspects malattia mitocondriale. Se the rispondere to ogni due delle tre rules of thumb è si, e le ragioni per these affirmative answers non sono spiegata by il paziente’s presenti diagnosi, poi a mitocondriale lavoro-up è giustificati. Tabella 2 elenca the standard esami che sono necessari in la valutazione of sospetta malattia mitocondriale. I call questo the “5 + 2” valutazione. Frequently, the provoca di queste esami will suggerire altro studi che può necessitare di essere condotte, ma these 7 esami will fornire a solid database upon con the razionale selezione di ulteriori studi |
Tabella 1. Rules of Thumb Think mitocondri quando: 1.A “comune malattia” ha atipiche caratteristiche che set it apart from the pack. 2.Three o più sistemi d'organo sono coinvolti. 3.Recurrent setbacks o flarei-nu ps a cronica malattia avvengono con infezioni. Tabella 2. Esaminazione diagnostica per Mitocondriale malattia 1.Sangue per mtDNA (PCR e Southern) 2.Sangue e CSF per Lattato e Piruvato, o MR Spectroscopy 3.Urine Organic Acids (by GC/MS) 4.Plasma e urine Amino Acids 5.Sangue e urine Carnitine 6.MRI del cervello 7.Muscle Biopsy Neuropathology e Electron Microscopy Mitocondriale Electron Transport Studi Fresh (coupled) mitocondriale Polarography Muscle mtDNA (PCR e Southern) può essere basato. La prima five esami elencato (omitting cervello spettroscopia per the moment) sono relativamente non-invasive e can solitamente be condotte per circa $1500. The last due esami sono più costly, ma sono considerate by molti specialisti di essere the più informative. The cervello MRI e biopsia muscolare, insieme con the associati catena respiratoria analisi, e mtDNA esaminazioni may costo circa $5000. When the provoca di queste sette studi sono combinate con attenta storia medica, famiglie storia, e serial fisica examinations, e assemblati by a metaboliche specialista con esperienza in malattie neurometaboliche e medicina mitocondriale, una diagnosi accurata può essere raggiunto in circa 50% del adulto pazienti riferite per sospetto di malattia mitocondriale. Fra bambini, the yield è più alte. These studi permettere una diagnosi accurata in circa 75% del bambini riferite per valutazione of sospetta malattia mitocondriale. The figures quoted in the paragraph sopra sono influenzato by the current state del art, e by the nature del pazienti riferite to a specialty centri per diagnosi. Research è being conducted today che will cambiano these figures significantly. New malattie sono being scoperta. New metodi of diagnosi sono being sviluppato. New experimental sistemi e animal modelli di malattia mitocondriale sono being constructed, e nuovi trattamenti sono |
Think Mitocondri 37 UMDF
| being tested. Entrambi clinica e basilari ricerche sono absolutely l'essenziale per questo procedere. The storia of scientifiche procedere ha taught us che the la maggior parte delle monumental discoveries non sono predictable. We cannot foresee what the next five anni sarà like per medicina mitocondriale, ma basato sulla current rate of crescita, è sicuro to say che ci sono still a numero of surprises in store, che molti cherished beliefs will fall, e nuovi ideas circa the ruolo e funzione dei mitocondri in umana malattia sarà espanse dramatically. Riferimenti 1. Barile, M; Valenti, D; Quagliariello, E; Passarella, S. Mitocondri come cellula targets of AZT (zidovudine). General Pharmacology, 1998 Oct, 31(4):531-8. 2. Christodoulou J. Somatic cellula mitocondriale mutazioni e catena respiratoria malattie. Presented a The Bottleneck Serono simposio, Sydney Australia, 7-8 Maggio 1999. 3. Damore, ME; Speiser, PW; Slonim, AE; New, MI; Shanske, S; Xia, W; Santorelli, FM; DiMauro, S. Early insorgenza of diabete mellito associati con the DNA mitocondriale T14709C point mutazione: paziente riportano e letteratura rassegna. Journal of Pediatrico Endocrinologia e Metabolism, 1999 Mar-Apr, 12(2):207-13. 4. DiMauro, S; Hirano, M. Mitocondri e malattia cardiaca. Current Opinion in cardiologia, 1998 Maggio, 13(3):190-7. 5. Fischel-Ghodsian, N. Mitocondriale sordità mutazioni reviewed. Human Mutation, 1999, 13(4):261-70. 6. Fiskum, G; Murphy, AN; Beal, MF. Mitocondri in neurodegenerazione: acute ischemia e cronica neurodegenerative malattie. Journal of Cerebrali Sangue Flow e Metabolism, 1999 Apr, 19(4):351-69. 7. Graff C, Clayton DA, Larsson N-G. Mitocondriale medicine—recente advances. J Internal Med 1999; 246:11-23. 8. Hatch, GM. La cardiolipina: biosintesi, rimodellazione e trafficking in the cuore e mammalian cellule (Rassegna). International Journal of Molecular Medicine, 1998 Jan, 1(1):33-41. |
9. Heales, SJ; Bola–os, JP; Stewart, VC; Brookes, PS; Land, JM; Clark, JB. Nitric ossido, mitocondri e neurologico malattia. Biochimica et Biophysica Acta, 1999 Feb 9, 1410(2):215-28. 10. Jansen, RP; de Boer, K. The bottleneck: mitocondriale imperatives in oogenesis e ovarian follicular fate. Molecular e Cellular Endocrinologia, 1998 Oct 25, 145(1-2):81-8. 11. Kerr, DS. Protean manifestazioni delle malattie mitocondriali: a mini rassegna. Journal of Pediatrico ematologia/Oncology, 1997 Jul- Aug, 19(4):279-86. 12. Kösel, S; Hofhaus, G; Maassen, A; Vieregge, P; Graeber, MB. Role dei mitocondri in Parkinson malattia. Biological Chemistry, 1999 Jul-Aug, 380(7-8):865-70. 13. Latkany P, Ciulla TA, Cacchillo PF, Malkoff MD. Mitocondriale maculopathy: geographic atrofia del macula in MELAS associati Q to G 3243 DNA mitocondriale point mutazione. Am J Ophthalmol 128:112-114, 1999. 14. Luft, R. Lo sviluppo of medicina mitocondriale. Proceedings del Nazionale Academy of Sciences del United States of America, 1994 Sep 13, 91(19):8731-8. 15. Luft, R; Landau, BR. Mitocondriale medicine. Journal of Internal Medicine, 1995 Nov, 238(5):405-21. 16. Niaudet, P; Rötig, A. Renale coinvolgimento in citopatie mitocondriali. Pediatrico nefrologia, 1996 Jun, 10(3):368-73. 17. Susin, SA; Zamzami, N; Kroemer, G. Mitocondri come regulators of apoptosi: doubt no più. Biochimica et Biophysica Acta, 1998 Aug 10, 1366(1-2):151-65. 18. Treem, WR; Sokol, RJ. Disorders del mitocondri. Seminars in Fegato malattia, 1998, 18(3):237-53. 19. Wallace D. DNA mitocondriale in invecchiamento e malattia. Sci Am 8(Aug):40-47, 1997. 20. Welch, KM; Ramadan, NM. Mitocondri, magnesium e emicranie. Journal del Neurological Sciences, 1995. |
UMDF 38 Think Mitocondri
Gestione Strategy per Acute Illness in i pazienti con
Mitocondriale Cytopathy
by Russell P. Saneto, DO, PhD, Pediatrico Epilessia
e Bruce H. Cohen, MD Pediatrico Neurology
The Cleveland Clinic Foundation, Cleveland, OH
© The Mitocondriale News, United Malattie mitocondriali Foundation, 2000
| Introduction The precarious health di un paziente con a citopatia mitocondriale rappresentano the fine line fra piccolo energia reserve e potenziale energia carenza. When domanda of aggiunto richiesta di energia avvengono, come they do in an acute affezioni, the diminuita reservoir of stored energia in un paziente con a citopatia mitocondriale spesso cannot compensate per the nuovi domanda di energia. When combinate con the diminuita inherit capacità to manufacture energia, il paziente’s bioenergetic health è alterata e a bioenergetic crisi può avvenire. Questo è specialmente vera in giovane bambini, che hanno piccolo energia reserves to begin con, e in those con a grave citopatia mitocondriale. Sebbene il numero of cose che può causare eccessivo bioenergetic sforzo è grande, we maggiormente vedi compromised bioenergetic health nel contesto di un'altra affezioni. I malesseri virali e fevers per esempio, può avere mitocondriale conseguenze. A quick e semplice rassegna della funzione mitocondriale è importante capire questo articolo. We consume cibo a fare the energia il nostro corpo needs to funzione. The energia in cibo è contained in the cleavable bonds fra the atoms in molecole of sugars (carboidrati), grassi, e proteine. Healthy mitocondri will generate 36 molecole di ATP (adenine trifosfato, ogni ATP rappresentano a unit dell'energia) per ogni molecule del glucosio che i mitocondri can burn o oxidize. Se i mitocondri do non funzione (con non è compatible con persino a breve vita di ogni persona), glucosio non è pienamente burned e solamente 2 molecole di ATP will essere prodotto. In questa situazione, c'è anche la produzione di due molecole of acido lattico. Studi della funzione mitocondriale in alcune of our sicker pazienti mostrano che sotto ideal laboratorio condizioni, solamente circa 40 – 60% del massimale energia può essere prodotto (14 - 21 molecole di ATP per ogni glucosio burned). Questo è an estimate of teorici produzione di ATP, con dovrebbe diminuire se laboratorio condizioni mimicked |
what avvengono nel corpo durante grave viral malesseri, disidratazione, e alto febbre. A utili analogy è to think di un otto-cylinder car che è running on solamente 6 o 7 cylinders. Come long come the car è on orizzontale ground the car’s prestazione can appare accettabile. Comunque, quando the domanda sono aumentato, tali come quando the car è loaded con passengers, attempts to climb a hill, o accelerates to entrare the freeway, there non è sufficiente energia prestazione to accomplish the task. The car knocks, sputters, e lags in
acceleration. Under these circostanze, the car |
Think Mitocondri 39 UMDF
| imbalance e possibili susseguente fisiologiche danno. There non è molto nella letteratura medica che è instructive in medicamente managing the crises of affezioni o altro stressful event in a paziente mitocondriale . What follows è basato on our sperimentano e understanding of alcune del pratici e teorici implicazioni of how il corpo’s biochimica colpiscono the bioenergetic health of a paziente mitocondriale . Preparation è the Key to Energetic Health C'è a ampia spettro of citopatie mitocondriali. Ciascuno uno è espressa in a unica way che è particolare to a specifico persona. Pertanto, non c'è “uno” meglio trattamento per those con a citopatia mitocondriale. Ciascuno paziente ha di essere cared per on una persona basis. Inoltre, our current understanding delle malattie mitocondriali è limitato e perciò, a meglio trattamento protocollo non deve exist. At presenti, ci sono molti più unknowns than provato trattamenti in the medica care di un paziente con a citopatia mitocondriale. The fragility of qualcuno con a citopatia mitocondriale richiede a lavorando knowledge of what segni to look per durante an acute affezioni. Per the scopi di questa discussione, we sarà discussing la gestione of fevers, incapacità consumare abbastanza liquid e cibo, e disidratazione. Alcuni pazienti vomito excessively, altri Hanno aumentato tremulousness, mentre still altri stop drinking e eating, e alcune Hanno cognitive cambiamenti. E' importante per the genitore o caregiver sapere these segni (Tabella 1 on page 39) per loro loved uno e call loro dottore se these segni sviluppare. In aggiunta, there devono essere a buon lavorando relationship con the primaria care medico, così quando the genitore o caregiver begins per vedere the segni of rapid bioenergetic decline, the medico can make arrangements per ospedalizzazione. A plan per tali eventi devono essere ben sviluppato by the medico prima to a crisi. Past experiences will dictate the necessitano per ospedalizzazione e the immediate trattamento once il paziente ha arrived a the ospedale. Unnecessary ospedalizzazione può avvenire on occasion, quando il paziente non è come sick come originally believed di essere, ma entrambi medico e caregivers quickly learn quando e quando non ospedalizzazione è necessaria. At la prima segni di un affezioni, the genitore o caregiver devono essere quick to implement trattamento. |
Questo generalmente comprendono liquidi e zucchero, e we trovano che alcune comune bevande per sportivi tali come Gatorade aiuto. High carboidrati pasti, dati by frequente feedings, anche possano aiutare replenish e sustain the necessaria livelli del glucosio per metabolismo. Per esempio, abbiamo usati aggiunto uncooked corn starch in un pasto per aumentare il livello del glucosio in the meal. L'adozione of medicamento to reduce febbre, tali come ibuprofen o acetaminophen devono essere usati. The dottore should calculate the proper dose di queste farmaci (10 – 15 mg/kg/dose dati ogni 4 – 6 ore), così che ci sono no fevers. Trattamento forWorsening Clinical Status E' difficile to pinpoint the time quando il paziente needs ospedalizzazione. Experience e comunicazione con the medico/health care team sono necessaria. Decisions può essere made quando the genitore/caregiver notices che, a dispetto the aggiunto misure of aumentato liquidi e extra carboidrati- contenente pasti/drinks, il paziente ha non risponde appropriately. Questo sarebbe più urgent se il paziente continui to peggiorare. Per ogni paziente, the dictating sintomi sono differenti, comunque, attraverso consulto con the health care team the appropriate decisione può essere made. Once the decisione è composta, il paziente e genitore/caregiver should go nel nearest ospedale. We tend to Hanno our pazienti admitted direttamente nel ospedale, ma questo will vary according to il paziente’s dottore. Our decisioni on what to do next sono basato on our knowledge of what deficit our paziente may Hanno. Once il paziente è stata checked into the ospedale, abbiamo a generale plan per proceeding ahead. La maggior parte dei pazienti will necessitano sangue e urine esami, placement di un IV, e liquidi iniziato. Laboratory Tests: The sottostanti ragione per the cambiano in bioenergetic status needs di essere addressed. Per esempio, se c'è una infezione, questo può necessitare di essere trattati. Se è asma, poi the proper respiratoria farmaci necessitano di essere dati. Laboratory esami può necessitare di essere ottenuto, comprendenti lattato, piruvato, ammoniaca, elettroliti e urine analisi. These valori may assist in understanding the depth of bioenergetic compromise. Per instance, se the lattato livello è alto, poi il quantitativo di destrosio |
UMDF 40 Think Mitocondri
| aggiunto nel bilanciata salt soluzione usati per idratazione può essere determinata. The livello of BUN will aiuto determine il livello of disidratazione e the rate of liquidi administration. Se le urine analisi indica che corpi chetonici sono being escrete nelle urine, questo è una indicazione che grassi sono being mobilizzate e maybe carnitina needs di essere aggiunto to i liquidi. Disidratazione: The grado of disidratazione è molto importante. Questo è perché disidratazione may adversely affect il cervello, muscoli, cuore, e reni. Persino lievi gradi of disidratazione, causato by vomito, diarrea, o febbre may grandemente limitare the reni’s capacità to get rid of a tossico metabolita, set the condizioni per rising metabolita livelli e induce ulteriori danno. Questo è the probabilmente meccanismo per the evolution of gangli basali danno in casi of metilmalonica aciduria e tipo I aciduria glutarica. We solitamente begin destrosio contenente a bilanciata salt soluzione, solitamente D5 o D10 con 1⁄4 o 1⁄2 normale saline (a salt mixture contenente 5% o 10% destrosio) e aggiunto carnitina. Dato una particolare situazione, il quantitativo of salt o zucchero potrebbero più alte o minore in the IV soluzione. The percentuale di destrosio contenente liquidi depends sulla anormalità del paziente. The rate a con liquidi è dati è individualizzato dipendendo sulla grado of disidratazione, e è lo stesso regardless of whether o non qualcuno ha una malattia mitocondriale. The normale criteri usati to decide whether to somministrare liquidi endovena devono essere abandoned in those con acute affezioni e disidratazione, come orale reidratazione terapia non deve offrono lo stesso grado of control e there non è come molto room per error in qualcuno da una malattia mitocondriale. Glucose: Why use aggiunto destrosio (glucosio) in a citopatia paziente mitocondriale che è dehydrated e/o ha acidosi lattica? Let’s use the automobile engine analogy di nuovo. In a citopatia paziente mitocondriale , the necessitano per fuel è più pronounced than in a normale paziente. Da quando the engine non deve funzione optimally, we necessitano to either aumentare the octane del fuel così the engine gets più output from the fuel o give the engine più fuel to burn. By giving il paziente più glucosio in intravenoso liquidi we sono accomplishing entrambi, più glucosio o fuel to burn e a più alte octane by |
migliorante the purity del fuel to burn, e di conseguenza produce più immediate energia (invece del grassi acidi from la demolizione dei grassi). By trattamento disidratazione, we sono anche producing an ambiente per the engine, con è migliore per energia efficienza. In più scientifiche termini, what we sono cercando to do è diminuire the acidosi lattica mentre expanding the volume of liquidi nel corpo. Lattato, ma anche altro tossine, può essere poisons to il cervello e come previously mentioned disidratazione can concentrate tossico metaboliti e diminuire the reni’s capacità to get rid di queste metaboliti. Lattato è the byproduct of inefficient glucosio metabolismo dovuta al disfunzione mitocondriale. When lattato builds up, it causa the sangue to diventata acidotic. The fegato, in a non-paziente mitocondriale , can utilizzano molto del lattato prodotto to remake glucosio per storage e anche burn it per fuel. Comunque, quando the pH ricade sotto a alcuni point, sotto 7.1, the fegato ceases usando lattato e invece provoca lattato. By giving liquidi, we sono expanding the volume del sangue e allowing the kidneys per aiutare rimuovere alcune del tossine. In aggiunta, the aggiunto liquidi è aiutanti the kidneys reverse the acidosi. Under condizioni of grave affezioni, è easier per il corpo to burn glucosio, piuttosto than grassi, per energia. The hopeful risultato of liquidi endovena con aggiunto glucosio e carnitina è the risoluzione of acidosi lattica, correction of electrolyte equilibrio, e the risoluzione dei sintomi. E' critiche to note che eccesso del glucosio può essere altamente tossico to a persona con piruvato deidrogenasi (PDH) carenza. In alcune situazioni of grave mitocondriale insufficienza, eccesso glucosio può provocare in peggioramento acidosi lattica come pure. In alcuni emergent casi, abbiamo had to add an insulina drip (0.03 unità/kg/hr – 0.1 unità/kg/hr) per aiutare migliora funzione mitocondriale by making glucosio più disponibili per i mitocondri e lowering libera grassi acido livelli, con possono migliorare the funzione of sick mitocondri. These sono molto selezionati casi, e consulto con a mitocondri esperti è necessaria per valutare e implement insulina in these special casi. Levo-Carnitine: In alcune pazienti, we will give a bolus of levo-carnitina (solitamente 50 mg/kg, seguite by 100 mg/kg/al giorno in 3 divided dosi) come questi pazienti solitamente presenti con a lattato acidosi e |
Think Mitocondri 41 UMDF
| Hanno begun to break down grassi into grassi acidi. In the analogy del car engine, quando the fuel è impropria per the engine ci sono by-prodotti prodotto che can diminuire la prestazione del engine. Uno can think of carnitina come una fuel additive per aiutare prevenire the build up of tossico by-prodotti created by inefficient fuel utilizzazione. Carnitine binds tossico-libera grassi acidi e acidi organici. In aggiunta, it agisce come una membrane mitocondriali stabilizer (seals the “leaky” gasket). Spesso pazienti con citopatie mitocondriali Hanno a secondari carnitina carenza come una risultato of overproduction of libera grassi acidi. The carnitina carenza sarebbe peggiorate by an acute insulto per i mitocondri e the metaboliche machinery. Aggiunto carnitina durante the acute stage di un affezioni dovrebbe aiuto in rimozione tossine e il miglioramento the carnitina carenza. Comunque, ci sono situazioni quando aggiunto carnitina può non essere necessaria o may potenzialmente peggiorare the situazione. Altro Integratori: Ci sono occasions quando altro integratori, in aggiunta a quelle sopra, può essere necessaria. We Hanno pazienti che hanno grave muscoli e periferico nerve compromissione quando affezioni anche induces cambiamenti in bioenergetic omeostasi, molto similare to cronica inflammatory demielinizzante polyneuropathy. Altro pazienti Hanno grave movimenti malattie, tali come distonia. We Hanno trovati che intravenoso gamma globulina (1 – 2 gm/kg in 1 o 2 dosi) temporarily migliora these condizioni. Sebbene non FDA approvato per queste malattie, abbiamo visto questo trattamento aiuto reverse neuropathic debolezza e dystonic movimenti. C'è alcune sperimentano con l'uso della creatina in pazienti avendo miopatie mitocondriali. We Hanno usati creatina come solamente a breve termine trattamento quando cercando per prevenire un paziente from being collocate on a respiratori macchina. Il corpo adatti to long termine use della creatina e presumably la sua efficacia lessens. We |
dovrebbe solamente raccomandiamo these tipi del trattamento in consulto con a mitocondri esperti. Conclusion Ci sono a few importante punti ricordare quando dealing con an intervening affezioni in a persona con a citopatia mitocondriale. 1. Il paziente o caregiver, insieme con the primaria care medico, should sviluppare a plan of how these malesseri sarà approached ahead di tempo. 2. In molti pazienti, c'è piccolo capacità per compensare durante an acute affezioni, così precoce interventi e l'uso of liquidi endovena sono spesso ricercato. 3. Once IV idratazione ha iniziato, it può essere necessarie to stop tutti attempts a feeding, allowing the intestino to riposo, fino a che il paziente begins to request liquidi o cibo. 4. Il miglioramento può essere slower than sarebbe expected in altrimenti persone sane, ma la maggior parte delle pazienti restart orale idratazione e feeding entro 12 – 24 ore dei liquidi endovena avendo begun. 5. The fonte di infezione devono essere sought, e se c'è a batteri infezione, it devono essere trattati con appropriate antibiotici. Viral infezioni non dovrebbero be trattati con antibiotici, come these do non lavoro contro viruses e molti antibiotici can ulteriori limitare funzione mitocondriale. Glossary of Termini Sugars: a generale termine usati per definire a semplice carboidrati. Glucose: un comune zucchero contained in sucrose (table zucchero) o lactose (latte zucchero). Dextrose è the pharmaceutical termine per glucosio. Bioenergetic Health: a descriptive termine usati per definire la capacità di produrre an adeguate supply dell'energia che will meet il corpo’s la domanda di energia. |
UMDF 42 Think Mitocondri
Anesthesia e Mitocondriale Citopatie
By Bruce H. Cohen, MD, John Shoffner, MD, Glenn DeBoer, MD
© The Mitocondriale News, United Malattie mitocondriali Foundation, 1998
| Introduction Questo articolo will outline alcune aspetti basilari of anestesia e address il problema del special rischi of anestesia in pazienti con citopatie mitocondriali. The applicability di queste raccomandazioni to una particolare paziente è complesso e devono essere individualizzato by il suo medico. Considerazioni comprendono whether il paziente non è diagnosticata (cioè receiving loro first valutazione per determinare whether una malattia mitocondriale è presenti), whether il paziente carries la diagnosi di una malattia mitocondriale, e what criteri were usati in making la diagnosi. The condizione clinica del paziente è probabilmente le più importanti aspetto del pre-operative valutazione. Alcuni pazienti Hanno minimal malattia manifestazioni e sono a basso rischio per complicazioni, laddove, altro pazienti Hanno significante malattia manifestazioni tali come debolezza dei muscoli respiratori, deglutizione difficoltà, malattia epatica, e malattia cardiaca, e sono a alto rischio per complicazioni. Anesthesia, spesso riferite to come anestesia generale, è the medica procedure che renders pazienti unconscious, insensibile al dolore e fornisce muscoli relaxation. Con anestesia locale, o regionale anestesia, pazienti sono awake o sedated, ma do non sentono dolore perché il dolore percorsi sono “blocked” by the anestetici locali. Spinal anestesia implica l'uso of anestetici locali o narcotici injected attorno the midollo spinale, causando loss of sensation sotto il livello che il medicamento è injected. Intravenous Anesthetics In ancient times orally somministrato extracts of poppy seeds (opium e morphine), extracts del deadly night shade (hyoscene e belladonna), e extracts of fermentations (alcool) were usati per “anestesia”. Tutti questi farmaci diminuire consciousness e the awareness di dolore. At presenti, la maggior parte di queste farmaci o loro modern counterparts sono somministrato intravenously. Thiopental è a rapidamente acting barbiturate usati per induction (la prima parte of anestesia quando the |
awake paziente è put to sonno) of anestesia. Propofol e etomidate sono nuovi rapidamente acting agenti induttori. Diazepam (Valium) e midazolam (Versed) sono farmaci in the benzodiazepine categoria, con sono potent hypnotics (induce a sonno-like state). Morphine, meperidine (Demerol) e fentanyl sono esempi of potent narcotic dolore relievers (analgesici) che sono usati come parte of alcune anestetici. Inhalation Anesthetics The earliest modern inhalation anestetici were the gas ossido nitroso (anche riferite to come laughing gas), e ether, a potent inhalational anestetici. Potent inhalational anestetici sono vapors prodotto da una liquid che evaporates facilmente. Nitrous ossido è still a basilari in modern anestesia; ma altro modern potent inhalational anestetici comprendono alotano, enflurane, isoflurane, sevoflurano, e desflurane. The potent agenti da inalazione anestetici fornire tutti gli modalità of generale anestesia, i quali comprendono unconsciousness, analgesia e lievi muscoli relaxation. Muscle Relaxants In aggiunta nel farmaci che induce the sonno-like state, un gruppo dei farmaci che fornisce muscoli relaxation sono spesso parte of a modern anestetici. These farmaci interfere con the comunicazione fra nerves e muscoli, e induce a paralyzed state così che il paziente non deve unconsciously move durante chirurgia. Ci sono di due tipi dei muscoli rilassanti. The depolarizzanti muscoli rilassanti (succinilcolina), cause il paziente’s muscoli muovere prima paralysis avvengono e sono relativamente short acting, mentre the non-depolarizzanti muscoli rilassanti do non cause tali movimenti. When pazienti sono paralyzed con questi farmaci, l'anestesista o anesthetist deve breathe per il paziente by either hooking the respiratori tube to a macchina o manually squeezing a “bag” spesso contenente a mixture di ossigeno, laughing gas e a potent inhalational anestetici. |
Think Mitocondri 43 UMDF
| Issues of Anesthesia in Mitocondriale Citopatie Modern generale anestesia consiste of induction con intravenoso agente e mantenimento con agenti da inalazione e/o con intravenoso agenti. Muscle rilassanti può o non può essere usati. Concerns circa the effetti collaterali e possibili complicazioni associati con chirurgia e anestesia sono shared dai pazienti con citopatie mitocondriali, loro famiglie e loro medici. The vast majority dei pazienti con citopatie mitocondriali Hanno an uneventful chirurgia e anestesia. i pazienti raramente sperimentano a complication con a semplice elective chirurgico procedure tali come una biopsia muscolare o gastrostomy tube placement. i pazienti con preoperative problemi respiratori sono a la più grande rischio per worse problemi dopo chirurgia. Similarmente, those con attacchi epilettici may sperimentano post-operative attacchi epilettici. Ci sono a limitato numero di rapporti descrivendo eventi dannosi e risultati in pazienti con malattie mitocondriali a seguito chirurgia e anestesia. Our knowledge circa these potenziale complicazioni sono basato on these anedotticamente rapporti. It non è possibili to draw conclusioni circa the sicurezza di una particolare anestetici agente basato sulla outcome di queste casi. Sebbene è possibili to esame una particolare anestetici in a laboratorio setting per vedere how it colpiscono funzione mitocondriale, questo lavoro è basato on animal experiments. How these animal studi relate to esseri umani in a clinica (non laboratorio) setting è impossibile per determinare. In reviewing these rapporti, a numero of inferences può essere made: • i pazienti con citopatie mitocondriali, on average, sono “sicker” than the unaffected paziente undergoing chirurgia e sono avendo an operative procedure per potenzialmente più seri ragioni than the unaffected paziente. • Come a generale rule, pazienti con citopatie mitocondriali sono a la più grande rischio than unaffected persone per effetti collaterali of alcune farmaci. Sebbene alcune farmaci may interfere con metabolismo energetico to alcune grado, complicazioni sono solitamente relativo nel condizione clinica del paziente prima to chirurgia. • The eventi dannosi riportati comprendono nuovi problemi neurologici tali a ictus, peggioramento del overall neurologici status, respiratoria difficoltà, attacchi epilettici, cardiaca aritmie, prolungato coma e morte. • Ipotonia (basso muscoli tono), disfunzione bulbare (debolezza dei muscoli che protegge the vie aeree) e |
relativamente scarsa ventilatory funzione (diminuita capacità to breathe deeply e tosse) sono comune in pazienti con malattie mitocondriali e pose an aumentato rischio per perioperative polmoniti. In uno studio dei pazienti con tipico sindrome di Leigh (con generalmente rappresentano uno del più grave forme of citopatie mitocondriali), respiratoria difficoltà prima to anestesia e chirurgia were a predictor of postoperative insufficienza respiratoria e morte. In the casi riportati, i pazienti awakened da anestesia ma deteriorated entro a al giorno. There did non seem di essere ogni specifico anestetici agente o tecnica che triggered these eventi dannosi. Non è chiaro from questo studio whether the deteriorazione was a diretta risultato del chirurgico procedure, the anestetici farmaci, o mitocondriale insufficienza dovuta al inadeguata ossigeno, derivanti da an unrecognized polmoniti o insufficienza respiratoria . Capire questi fattori may make anestesia safer, ma will non evitare tutti rischi. (J Child Neurol 1990;5:137-41) • Malignant Hyperthermia (MH) è a vita threatening, ereditata sindrome triggered by potent agenti da inalazione anestetici e/o depolarizzanti muscoli rilassanti. E' causato by anormale aumento in muscoli calcio concentrazioni portante a non controllate muscoli metabolismo, susseguente acidosi metabolica, muscoli danno, e elevati potassio livelli.Senza trattamento, MH will spesso risultato in morte. i pazienti a rischio per MH possono svilupparsi il disturbo con loro first anestetici, o may Hanno a dozen o più anestetici senza a problema, solamente to sviluppare MH con the next anestetici. Ci sono specifico trattamenti disponibili per MH se it should sviluppare, ma the meglio approccio è to identificare pazienti a rischio e use anestetici che do non trigger MH. Rischio fattori per MH comprendono 1) prima MH episodio nel paziente, 2) a famiglie storia of MH e 3) muscoli malattia. Molti pazienti con citopatie mitocondriali talvolta Hanno an associati miopatia (muscoli malattia), con places them a potenziale rischio per MH. Ci sono anestetici che sono “sicuro” per those con o a rischio per MH, ma these anestetici may adversely affect funzione mitocondriale in alcune pazienti. • The rischio of insufficienza respiratoria e peggioramento of neurologici funzione è spesso notati in pazienti con citopatie mitocondriali dopo “stressful” malesseri, comprendenti infezioni tali come routine viral malesseri o polmoniti. Infezioni may risultato da una complicaThink |
UMDF 44 Think Mitocondri
| tion of chirurgia o from the necessitano per chirurgia, come nella il caso of a ruptured appendix. Infezioni, tali come the comune freddo, può anche avvengono randomly attorno the time of chirurgia. Certamente chirurgia stessa, persino se per a non-emergency condizione, è a maggiore sforzo. The a seguito discussione è abbastanza complicata ma necessarie in order capire che anestetici farmaci alone non dovrebbero be considerate the solamente element in portante a these adverse risultati. Durante infezioni, il corpo risponde by making sostanze chimiche conosciuta come cytokines. Cytokines aiuto il corpo fight infezione, e sono anche responsabile per the febbre, aches, chills e the overall “rotten” feeling we get quando we sono ill. Cytokines induce la formazione of nitric ossido. Nitric ossido (the chimiche formula è NO) è a potente ossidante con molti utili scopi in our corpi, alcune of con seem abbastanza unrelated, tali come forming nuovi memories e killing batteri. Comunque, nitric ossido inibisca cis-acotinase (a ciclo dell'acido citrico enzimi) e the ferro-contenente cytochromes della catena respiratoria. Pertanto, NO in alto amounts può diminuire produzione di energia, con è ill-afforded in pazienti who already Hanno an danneggiato capacità per generare energia. Nitric ossido può anche interact con altro sostanze chimiche nel corpo che risultato in danno nel DNA mitocondriale e mitocondriale structure stessa. TNF (tumore necrosis fattore) è uno cytokine che è conosciuta di essere released by il corpo durante chirurgia, e è anche conosciuta di essere a potent inhibitor of complesso III. TNF ha molti l'essenziale funzioni, e serve come una naturale la difesa contro infezioni e cancro. In altrimenti persone sane, the inhibitory effetto on complesso III è obviously non pericoloso, ma possono giocare alcune ruolo in persone con malattie mitocondriali, who non sono in grado a tollerare ogni piccole decrement in funzione mitocondriale. Pertanto, agenti anestetici può non essere responsabile, a meno senza ulteriori fattori, per causando neurologici deteriorazione. Entrambi the sforzo of chirurgia come pure ogni associati infezioni may trigger gli eventi portante a a deteriorazione in suscettibili pazienti. (Anesthesiology 1997;87:420-5) • i pazienti con cuore ritmo problemi, tali come those con Kearn-Sayres, sono a rischio per grave cuore elettrici conduzione blocks, la quale può portare a morte. Isoflurane può essere a preferito agente da inalazione all'opposto to Halothane, perché Isoflurane causa meno disturbi in cuore rhythms. (Anesthesiology 1994;49:876-878) • Sebbene spinal anestesia è sicuro, it devono essere |
usati con extra cautela in pazienti con neuropatie o miopatie, perché del possibili deleteri effetti on pressione sanguigna e respiratoria funzione. Raccomandazioni Non ci sono doubt che pazienti con una malattia mitocondriale can sottoporsi generale anestesia safely, come dimostrato by untold migliaia of uneventful surgeries e anestetici esposizione. La domanda che pazienti e loro medici wish sapere è how to ulteriori diminuire il rischio. The a seguito raccomandazioni sono made con the understanding che c'è piccolo dati suggesting che ogni specifico precauzioni can minore il rischio of neurologici eventi. Comunque, these raccomandazioni seem di essere prudente dati quale è conosciuta circa gli effetti di stress chirurgico, infezioni e agenti anestetici in pazienti con citopatie mitocondriali: 1. Strict attention dovrebbero essere fatto to respiratoria funzione prima, durante e dopo chirurgia, specialmente in pazienti con anormale preoperative respiratoria segni e sintomi. Vigorous respiratoria physiotherapy devono essere standard postoperative care in ogni paziente con polmonari difficoltà. Early use of artificial ventilazione, mantenendo normale oxygenation, CO2 eliminazione, e vigoroso respiratoria physiotherapy sono raccomandato a la prima sign of respiratoria deteriorazione. 2. There devono essere a heightened livello of sospetto per infezioni tali come polmoniti, con devono essere prontamente trattati. 3. Lactated Ringer’s soluzione (anche conosciuta come Ringer’s Lattato) devono essere evitati come un intravenoso liquidi, come it contains acido lattico. 4. Normal glucosio ematico, corpo temperature, e acido-base equilibrio devono essere mantenuti durante chirurgia. Low glucosio ematico devono essere evitati. Comunque, a alto glucosio ematico possono indicare an acute disturbazione in metabolismo del piruvato o fosforilazione ossidativa. In questa situazione, the acido lattico livelli may anche be elevati. 5. Evitare depolarizzanti muscoli rilassanti, sebbene queste sono state usati safely in molti pazienti con malattie mitocondriali. (Anesthesiology 1979;51:343-345.) 6. Delay elective chirurgia se c'è ogni evidenze di infezione. 7. Potent agenti da inalazione anestetici appare di essere |
Mitocondri 45 UMDF
| sicuro nella maggioranza delle persone con malattie mitocondriali. In pazienti a rischio per MH, tali come quei pazienti con miopatie che sono spesso associati con loro malattia mitocondriale, i rischi e alternative metodi of anestesia devono essere considerate by the medico. Certamente se c'è stata a previous adverse reazione nel paziente o famiglie membro, these agenti devono essere evitati. (Tabella 1) 8. Anesthesia con combinazioni of barbiturici, narcotici, benzodiazepine, e ossido nitroso anche pose a teorici rischio per pazienti con malattie della fosforilazione ossidativa. (Tabella 2) Questo rischio devono essere considerate solamente come una potenziale rischio a meno che un paziente ha con esperienza a bad reazione to ogni del farmaci. Questo apparente paradosso fra the due metodi of generale anestesia devono essere addressed con ogni paziente, e l'anestesista deve determine quale è the safest route. 9. Studi su animali indicate che propofol, un nuovo intravenoso anestetici, danneggia funzione mitocondriale to a la più grande grado than altro anestetici. Comunque, questo farmaco è stata usati safely come un |
anestetici in molti pazienti con citopatie mitocondriali. Ci sono state observations che prolungato continua use (giorni) a alto dosaggi per trattare frequente attacchi epilettici causa a sindrome similare to mitocondriale insufficienza, e di conseguenza l'uso prolungato in un paziente con a citopatia mitocondriale può non essere sicuro. Conclusion An aumentato awareness è necessaria whenever a persona con a citopatia mitocondriale è contemplating o undergoing a chirurgico procedure. By virtue del affezioni stessa, ci sono la più grande rischi coinvolti con ogni medica interventi. The safest anestetici non è conosciuta e the scelta di anestetici devono essere individualizzato to il paziente’s particolare needs. Sebbene agenti anestetici possono giocare a contribuire fattore in causando an adverse event associati con chirurgia, the affezioni, the sforzo of che affezioni, the chirurgico procedure e concurrent infezioni possono giocare a larger ruolo in causando neurologici deteriorazione.Con ulteriori ricerche, più sarà learned circa these problem |
Tabella 1: Malignant Hyperthermia (MH) Precauzioni
Factor Trattamento
History Acknowledge la potenziale per problemi in
pazienti con muscoli malattia, those con a past
storia o a famiglie storia of MH.
Muscle Relaxants Evitare depolarizzanti farmaci tali come
succinilcolina; e use non-depolarizzanti
agenti tali come pancuronium invece.
Anesthetic Agent Evitare the potent agenti da inalazione tali come
alotano e enthrane. Use agenti tali come
ossido nitroso, barbiturici, benzodiazepine,
e narcotici.
Preparation per MH Have adeguate amounts of Dantrolene®
disponibili e use it come appena come la prima segni of
MH avvengono.
Tabella 2: Effects of Anesthetic Agents of Mitocondriale Function
Medication Biochemical e Clinical Effects on
Mitocondriale Function
Barbiturates Inhibits Complesso I attività a alto livelli.
Benzodiazepines Inhibits adenosina nucleotide traslocasi
Propofol e/o lipidi carrier Inhibits funzione mitocondriale.
Halothane L'aumentata rischio per cuore disturbazione del ritmo.
Nitrous Oxide (chimiche formula è N2O) Neurotoxic, possibilmente by incrementare
nitric ossido
produzione, con inibisca cis-acotinase e
ferro-contenente trasporto degli elettroni enzimi;
colpente produzione di energia.
Non-depolarizzanti Agents Increasing sensibilità nel paralitico effetti
e prolungato risposte riportati.
Local Anesthetics Bupivacaine disaccoppia ossidazione e
fosforilazione.
UMDF 46 Think Mitocondri
sindrome di Leigh: Clinical Caratteristiche e
Biochemical e DNA Le anormalità
by David R. Thorburn, PhD
Royal bambini’s Hospital, Melbourne, Australia
© The Mitocondriale News, United Malattie mitocondriali Foundation, 1998
| Introduction sindrome di Leigh o malattia di Leigh è a degenerative malattia della fanciullezza, e è uno del più comune e meglio conosciuta del malattie mitocondriali. Inizialmente the bambino appears di essere growing normalmente e possono svilupparsi the usual precoce skills tali come smiling interactively, rolling over, pulling stessi up, talking e walking. Suddenly (spesso a seguito a viral infezione) o talvolta gradualmente, the bambino begins to lose these skills (ie regressione evolutiva). Questo loss of skills tipicamente diventa apparente fra 4 e 12 mesi di età, ma it può essere molto earlier o later, e occasionale pazienti apparentemente don’t show sintomi fino a che adulthood. The clinica sintomi vary fra bambini, ma comune caratteristiche comprendono an anormale respiratori ritmo (panting o sighing), clumsy o jerky movimenti e either wobbley o congelato eyeballs. La malattia tipicamente progresses ma a a variabile rate, spesso con sharp declines seguite by plateaus nelle quali the bambino may rimane relativamente ben e persino recover alcune lost skills, ma solitamente seguite by un'altra decline. We Hanno no cure per malattia di Leigh, sebbene alcune pazienti do show a parziale risposte to farmaco o dieta terapia. The UMDF asked me recentemente per permission da usare a scientifiche articolo (Rahman e colleghi, 1996, Annals of Neurology (1996) volume 39, pages 343-351) circa malattia di Leigh on con I was the senior author. Sfortunatamente, come con la maggior parte delle pubblicazioni, the copyright belongs nel publishing compagnie piuttosto than nel originale autori. Da quando the permission was non mine to give, I offered to write a summary, e attempt to rimuovere la maggior parte delle del scientifiche jargon così it sarebbe a piccolo più facilmente understood by the non-specialista. Ho avuto to leave in abbastanza a lot of dettaglio sebbene, e I indicherebbe reading the Introduction del lead articolo by Dr. Bruce Cohen in the Fall 1997 problema of UMDF Mitocondriale News per più sfondo sulla malattia mitocondriale. Our 1996 articolo was basically a summary of my laboratorio’s sperimentano |
i.of malattia di Leigh, e an attempt to rispondere questioni tali come: In how molti pazienti can we identificare a basilari biochimica difetto? How comune è ogni difetto? Do pazienti con differenti basilari difetti Hanno differenti caratteristiche cliniche? How comune è malattia di Leigh? What does tutti questo tell us circa the genetica of malattia di Leigh? We reasoned che pulling insieme what we knew del basilari biochimica e genetica dovrebbe migliora our capacità to sceglino razionale terapia per pazienti e to give accurate genetica counselling e prenatal diagnosi. A Brief History of malattia di Leigh The caratteristiche caratteristica of malattia di Leigh è the presenza of distinctive cambiamenti o lesioni in alcuni parti del cervello. Perciò fino a che recentemente, la diagnosi of malattia di Leigh could solamente essere fatto dopo morte. Questo neuropathology was first descritte come “Subacute Necrotizing Encephalomyelopathy” by a British pediatrician, Dr. Denis Leigh in 1951, e SNE è un'altra name talvolta usati per malattia di Leigh. Da quando Dr. Leigh pronounced his name in the English way, the correct pronunciation of malattia di Leigh è to rhyme con “Lee” piuttosto than “Lay”. Le lesioni cerebrali in malattia di Leigh were noticed di essere similare a quelle trovati in a malattia chiamati Wernicke’s encefalopatia. Dietary carenza di vitamine B1 (tiamina) contributes to Wernicke’s encefalopatia, e così precoce studi of malattia di Leigh concentrated on tiamina metabolismo, e it was consigliati che urine from pazienti contained a chimiche che blocked activation di tiamina. Altro studi attorno questo time (the 1960s) consigliati due altro anormalità in malattia di Leigh. Sangue from pazienti was trovati to Hanno grande amounts of acido lattico, a chimiche che tends to accumulate quando il corpo cannot generate energia normalmente from fuels tali come zucchero. Secondo, it was consigliati che una particolare enzimi (ie a proteine catalyst necessaria to convert uno chimiche into a differenti forma) nominato piruvato carbossilasi was |
Think Mitocondri 47 UMDF
| missing in alcune pazienti. In hindsight, alcune del precoce rapporti sulla roles of a tiamina inhibitor e carenza di piruvato carbossilasi in malattia di Leigh were probabilmente mistaken, dovuta al problemi con the metodi of esaminazioni disponibili a che time. Durante the 1970s comunque, it became chiara che alcune casi of malattia di Leigh were causato by ereditata difetti in due altro enzimi coinvolti nel metabolismo energetico, namely the piruvato deidrogenasi complesso (PDHC) e citocromo c ossidasi (COX). Figura 1 mostrano how these enzimi sono coinvolti in generating a chimiche chiamati ATP from sugars like glucosio. ATP è the chiave energia carrier che tutti our cellule necessitano, whether è usati per il nostro corpo to grow, our muscoli to contract, our cuore to pump, our cervello cellule to communicate, o our pancreas a fare e excrete insulina. COX è the quattroth componente of our centrale energia generating percorso, la catena respiratoria mitocondriale, e è anche conosciuta come complesso IV. The termini “catena di trasporto degli elettroni” e “fosforilazione ossidativa” o “OXPHOS” mean molto lo stesso thing come catena respiratoria.
|
Durante the 1980s, occasionale pazienti were riconosciuto con difetti of catena respiratoria complesso I (anche conosciuta come NADH deidrogenasi). We anche inizia rendersi conto the amazing complessità della catena respiratoria, the normale funzione of con richiede più than 100 differenti geni di essere lavorando appropriatamente. It became apparente che alcune malattie della catena respiratoria were non dovuta al mutazioni (o cambiamenti) in i geni del cellula nuclei, ma to mutazioni in the DNA mitocondriale (mtDNA). The mtDNA è presenti in migliaia of copies in ogni cellula inside our cellulari power plants, i mitocondri, con we inherit from our madre. In the precoce 1990s, mutazioni del mtDNA were shown di essere responsabile per alcune casi of malattia di Leigh. When uno considers how long it ha taken to lavoro out the genetica di malattie like cystic fibrosis e Huntington malattia, con entrambi coinvolgono just una singola gene, it non è surprising che procedere sembri lento con malattie della catena respiratoria mitocondriale. Our Survey of malattia di Leigh In 1992, I had the buon fortune to take over a laboratorio con had been iniziato by Dr. Garry Brown (ora a Oxford University). It had been la principale australiano referral laboratorio per bambini sospetta di malattie of generazione di energia, e had a raccolta of skin cellula campioni (o fibroblasti) dating back nel mid-1970s. Dato the recente rilevamenti del mutazioni del mtDNA in malattia di Leigh, I was keen per determinare how molti of our pazienti carried tali mutazioni. Questo was possibili thanks to a British pediatrician, Dr. Shamima Rahman, who spent a anno in Melbourne on an programma di scambio. She e I decided to look per mutazioni del mtDNA in tutti i pazienti we had been riferite per investigazione of malattia di Leigh. She anche reviewed tutti the medica records e cervello imaging studi di ogni bambino. Un altro importante aspetto of our studio was che prima to 1994, we (e la maggior parte delle altro laboratori) had non been in grado per misurare catena respiratoria complesso I in skin cellule. Thanks to a conversation a a scientifiche conference, we became in grado to do questo by modificanti a metodi usati in Dr. Doug Turnbull’s laboratorio in Newcastle, UK, abilitando us per diagnosticare carenza del complesso I in an ulteriori 9 pazienti. A definite diagnosi of malattia di Leigh è solamente |
UMDF 48 Think Mitocondri
| possibili se the bambino ha had cervello imaging (CAT scansione o MRI) o autopsy studi mostranti the tipico cervello cambiamenti. The gruppo of 67 bambini we studiati inclusi 35 who had a definite diagnosi, e 32 simil-Leigh bambini who had clinica sintomi suggesting malattia di Leigh ma who had non been shown to Hanno the tipico cervello cambiamenti. Alcuni di queste 32 quasi certamente had malattia di Leigh, ma had non had imaging o autopsy condotte. On average, these simil-Leigh bambini were milder than those con malattia di Leigh cioè, they lived più a lungo. The a seguito sections describe the differenti basilari difetti we trovati in our pazienti. How Common Is Ciascuno Basic Defect? mtDNA ATPasi6 Le mutazioni Our mtDNA è a circular genome coerente of 16,569 base appaia o nucleotides, con we numero from 1 to 16,569. Se an mtDNA mutazione colpiscono just uno nucleotide (ie a point mutazione), we refer to it by its numero nel mtDNA sequenza. Ten bambini had mutazioni a nucleotide 8993, nelle quali the normale T base had been cambiata to either a G o a C. The TG mutazione was first descritte in an mtDNA sindrome conosciuta come NARP, che implica Neuropatia (debolezza muscolare), Atassia (a movimenti disturbo) e Retinite Pigmentosa (a forma di cecità), e uno studio from NewYork had trovati previously che it was a relativamente comune cause of malattia di Leigh. The 8993 mutazioni sono in the Gene ATPasi6 del mtDNA con codifica parte del ATP sintetasi (complesso V) che forme the final parte del energia generating percorso. E' difficile per misurare complesso V attività by enzimi analisi, e così none of our 10 pazienti had had an enzimi diagnosi made previously.We di conseguenza sequenced the ATPasi 6 gene in 4 pazienti (e successivamente in a ulteriori 10) ma trovati no altro mutazioni in questo gene. Altro mtDNA Le mutazioni Uno bambino had a grande deletion che removed circa a quarter of loro mtDNA genome, e a second had a mutazione a nucleotide 8344, con è anche talvolta trovati in un'altra mtDNA sindrome, conosciuta come MERRF, a forma of epilessia. Questo mutazione è in the tRNA-Lysine gene, con insieme con the 21 altro tRNA geni è necessaria per la sintesi delle proteine mitocondriali ie a fare the proteine codificato by the altro mtDNA geni tali come |
ATPasi6. Perciò la maggior parte delle pazienti con mutazioni del tRNA anche Hanno an enzimi difetto of uno o più della catena respiratoria enzimi. Respiratory Chain Enzyme I difetti Le più comune basilari difetto in our gruppo dei pazienti was carenza del complesso I, colpente 13 of our 67 bambini. Questo enzimi difetto è stata underdiagnosed in passato perché the enzimi assay was problematici, Particolarmente in congelato muscoli e in skin cellule. Fino a che recentemente, forse solamente uno laboratorio (Dr. Brian Robinson in Toronto) had been in grado per misurare complesso I reliably in skin cellule, e ha avuto been the solamente uno descrivere più than uno o due casi of malattia di Leigh con carenza del complesso I. The disponibilità di nuovi sintetica substrati per the assay, e nuovi protocolli significa che it can ora be diagnosticata reliably. Nine altro bambini had carenza di un'altra catena respiratoria enzimi, namely complesso IV (COX). Piruvato Deyhdrogenase I difetti Seven bambini had carenza di PDHC. Seven differenti geni sono necessarie per normale PDHC funzione, ma uno di queste è a “hotspot” per mutazioni, namely the E1alpha subunità gene, con è trovati sulla X chromosome. The altro PDHC geni sono “autosomico”, ie they non sono presenti sulla X o Y chromosomes che determine our gender, ma on alcune del altro 22 chromosomes. Six of our PDHC pazienti were boys, e tutti sei had mutazioni in the E1alpha subunità gene. Questo gene appeared normale in the solamente girl con carenza di PDHC, suggesting Ha avuto a mutazione in uno del sei autosomico geni per PDHC. In How Molti i pazienti CanWe Identify A Basic Defect? In our studio, 80% del pazienti con definite malattia di Leigh, e 41% del simil-Leigh bambini had a basilari difetto identificata. Shortly dopo our studio, Morris e colleghi (Annals of Neurology, 1996, volume 40, pages 25-30) riportati a similare survey of British malattia di Leigh pazienti. They classificate pazienti come definite (pathologically provato) e probabile, e identificata a difetto in 74% del definite famiglie, e 39% del probabile famiglie. They anche confermata che carenza del complesso I was un comune diagnosi. In |
Think Mitocondri 49 UMDF
Frequenza of Recognised Basic I difetti in malattia di Leigh
Total Group
Molecular Defect
malattia di Leigh (35
pazienti)
‘simil-Leigh’
malattia (32
pazienti)
Number dei pazienti
(67 in tutti)
Number of
Families
(56 in tutti)
Male:Female
Ratio
Ereditarietà
mtDNA 8993 TG 4 2 6 6 3:3 Materna
mtDNA 8993 TC 2 2 4 3 4:0 Materna
mtDNA 8344 1 0 1 1 1:0 Materna
delezione nel mtDNA 0 1 1 1 0:1 Solitamente non
ereditata
PDHC 4 3 7 6 6:1 collegata a X o
autosomica recessiva
Complesso I 12 1 13 11 8:5 Autosomica
recessive, materna
o collegata a X
Complesso IV (COX) 5 4 9 7 6:3 Solitamente autosomica recessiva
i pazienti con difetto
(% con difetto)
28 (80%) 13 (41%) 41 (61%) 35 (63%) 28:13
| lo stesso problema of Annals of Neurology (pages 5-6) c'è una eccellente editorial by Dr.s DiMauro e De Vivo, che forniscono una panoramica del genetica of malattia di Leigh. E' ben riconosciuto che molti pazienti Hanno difetti che sono solamente rintracciabili in alcune tessuti (eg muscoli o cervello) e non in skin cellule. In our retrospettiva survey, skin cellule were the solamente campioni disponibili from due thirds del pazienti. Solo 52% di queste pazienti had difetti identificata, comparata con 76% of those from whom muscoli e/o fegato were studiati. Uno paziente had normale attività enzimatica in muscoli, ma the difetto was apparente in fegato. Perciò our sperimentano è che muscoli e/o biopsia epatica fornire the meglio chance of achieving a diagnosi in malattia di Leigh. Complesso I carenza e mutazioni del mtDNA sono state underdiagnosed in passato e insieme rappresenta the basilari difetto in più than uno-third of our totale gruppo (25 of 67 bambini). Clinical Caratteristiche In The Different Basic I difetti E' stata consigliati che caratteristiche cliniche tali come età of insorgenza e attacchi epilettici differ sostanzialmente fra pazienti con malattia di Leigh dovuta al differenti difetti. Sebbene we trovati alcune caratteristiche tended di essere più comune con differenti difetti, we trovati no striking differenze eccetto che pazienti con the mtDNA 8993 TC mutazione had |
later insorgenza e slower progressione than altro pazienti. The retrospettiva nature e size of our studio mean che we cannot esclude che alcune caratteristiche sono più comune in differenti gruppi eg retinite pigmentosa probabilmente è più comune con the 8993 TG mutazione, ma non tutti our pazienti had the proper tipo of esaminazioni (elettroretinografia) di essere alcuni. In generale comunque, it appears che specifico caratteristiche cliniche non sono strongly predictive del basilari difetto How Common Is malattia di Leigh? Per a numero of historical e geografico ragioni, we pensarono che our laboratorio knew of virtualmente ogni caso of sospetta malattia di Leigh in South Eastern Australia fra 1980 e 1992. Nobody had ever stimato how comune malattia di Leigh was, così we sperimentato to do questo. To be honest, questo was a the suggestion of a aiuto reviewer, piuttosto than our own idea. Durante questo periodo, we identificata 28 bambini con malattia di Leigh e 26 simil-Leigh casi. There were 2.2 milioni births durante questo periodo, con significa uno caso of malattia di Leigh per 77,000 births, o uno per 40,000 per Leigh e simil-Leigh malattia. These figures sono probabilmente di essere underestimates, since alcune bambini were probabilmente mai riferite to an appropriate neurologist o medica geneticist, e alcune wouldn’t Hanno had the necessarie indagini |
UMDF 50 Think Mitocondri
| (tali come MRI) per a definite diagnosi. We felt che the figure of 1 per 40,000 births was a ragionevole estimate per l'incidenza of malattia di Leigh. Questa figura dovrebbe imply che circa 100 nuovi casi devono essere diagnosticata ogni anno negli USA. The genetica Of malattia di Leigh The genetica of malattia di Leigh sono molto complesso ma the usual modes dell'ereditarietà di ogni basilari difetto sono riassunte in the Tabella. Up fino a che the precoce 1990s, it had been assumed che malattia di Leigh was nearly sempre an autosomica recessiva condizione. Autosomica recessive ereditarietà implies che the bambino ha ereditata a faulty copia of un gene nucleare from ogni genitore. All of us carry 5 to 10 tali faulty gene copies che sono harmless, a meno che our partner happens to anche carry a faulty copia of lo stesso gene. Se che è il caso, poi ogni gravidanza è a a 1 in 4 rischio del due faulty gene copies coming insieme, derivante in an affetto bambino. Perhaps the maggiore rilevamenti of our studio was che the assumption of autosomica recessiva ereditarietà dovrebbe sono state wrong in nearly uno metà del famiglie nelle quali a basilari difetto was identificata. E' stata conosciuta per molti anni che per ogni due girls con malattia di Leigh, circa tre boys sono affetto, e our studio confermata questo. Le mutazioni in geni sulla X chromosome tend to affect più boys than girls, since boys Hanno solamente uno X chromosome, e the second copia in girls fornisce alcune protection. carenza di PDHC causa alcune di queste eccesso of affetto boys, ma non tutti of it. A complesso I gene ha anche recentemente been shown di essere sulla X chromosome, ma altro fattori sono probabilmente anche coinvolti, con potrebbero ormonale, ambientale o genetica. E' chiara che mutazioni in vari parti del produzione di energia sistema può causare malattia di Leigh. Altro pazienti con difetti in lo stesso componenti may die appena dopo birth o sviluppare a differenti gamma dei sintomi in fanciullezza o adulthood. Questo suggerisce che malattia di Leigh è due più nel grado of compromissione della produzione di energia in alcuni regioni del cervello than nel particolare gene coinvolti. Perciò |
we view malattia di Leigh come parte of a spettro of generazione di energia malattie (vedi Figure 2). The fattori coinvolti in determinazione quando ogni paziente fits in questo spettro possono includere genetica fattori tali come the gene coinvolti, the particolare mutazione, the mutante load (per mutazioni del mtDNA), the X-chromosome inactivation modello (in girls) e ambientale fattori comprendenti infezioni e dieta. An Update sulla Cause of Leigh o simil-Leigh malattia Da quando our articolo was sottoposti, a numero of altro tipi of basilari difetto sono state riportati in malattia di Leigh. Questo comprende: altro mutazioni in the Gene ATPasi6; altro mutazioni del mtDNA in the tRNA geni per valina e triptofano; deplezione nel mtDNA (con significa non abbastanza copies del mtDNA, in contrasto to delezione nel mtDNA con significa normale numeri of mtDNAs ma alcune copies avendo a grande chunk missing) ; altro difetti della catena respiratoria enzimatici tali come carenza del complesso II e combinate complesso I e IV carenza. Sommario Of Major Ritrovamenti We were in grado to trovano the basilari difetto in circa 80% of bambini con malattia di Leigh, e 40% of bambini con simil-Leigh malattia. All la principale causa affect generazione di energia, e we confermata che carenza del complesso I e mutazioni del mtDNA (insieme con complesso IV e carenza di PDHC) sono comune anormalità. At meno tre differenti tipi dell'ereditarietà sono comune, così accurate genetica counselling can solamente be offered to a famiglie se the basilari difetto è identificata. Research advances durante the last decade mean che maggiore specialista centres sono ora in grado to identificare the basilari problema nella maggioranza dei bambini con malattia di Leigh. We Hanno anche migliorata our capacità offrire prenatal diagnosi in alcune famiglie to evitare avendo ulteriori affetto bambini. Chiaramente, the sfida è to build on questo ricerche base, offrire migliorata dieta e farmaco terapia e to sviluppare nuovi trattamenti tali come gene terapia. |
Think Mitocondri 51 UMDF
Ictus e Transient Events in Mitocondriale Citopatie
By: Bruce H. Cohen, MD
© The Mitocondriale News, United Malattie mitocondriali Foundation, 1998
| Introduction To the estesa che medici understand malattia mitocondriale, il cervello è visionate come the più vulnerabili organo, entrambi perché è spesso affetto e anche perché ogni cervello danno può essere catastrofica. La maggior parte dovrebbe agree che cervello disfunzione occurring in malattie mitocondriali colpiscono the qualità della vita più than la disfunzione of altro organi sistemi. Se it be ritardo mentale, attacchi epilettici o ictus, cervello disfunzione è ovvia. Uno dei più comune neurologici eventi comunemente associati con citopatie mitocondriali sono ictus. Nonostante questo associazione, ictus sono apparentemente meno comune in those con citopatie mitocondriali than altro problemi neurologici tali come attacchi epilettici, debolezza o cognitive ritardo. The vera incidence of ictus non è conosciuta. From a historical perspective, ictus were conosciuta di essere parte delle malattie mitocondriali. Uno del earliest malattie mitocondriali went by the acronym della MELAS (Mitocondriale miopatia, Encefalopatia, acidosi lattica e Ictus-Like episodi ), con è still uno del più comunemente descritte combinazione dei sintomi. Pressocché by definizione, those con MELAS soffrono ictus o “simil-ictus” eventi che seem like a ictus a first, ma quando il paziente returns to normale (o loro neurologici ‘baseline’) quickly. In aggiunta to ictus(s), those con MELAS anche Hanno vario gradi of acidosi lattica, come pure compromissione cognitiva, debolezza, attacchi epilettici e altro problemi neurologici. MELAS sarà discusse in la più grande dettaglio later in questo articolo. The terminology delle malattie mitocondriali non è facile capire. Nel passato, l'intero gruppo di malattie were spesso riferite to come mitocondriale encephalomyopathies, referring to malattie (pathy) colpente il cervello (encephalo) e muscoli (myo). Comunque, we ora understand che molti organi altro than il cervello e muscoli were affetto (Tabella 1) e di conseguenza il termine ‘citopatia mitocondriale’ è ora considerate the accettato designation. |
Brief Historical Rassegna E Basics Of Mitocondriale genetica E' importante capire termini ‘genotipo’ e ‘fenotipo’ quando discussing malattie mitocondriali. Questo concetto è importante capire perché it non è sempre possibili, usando current technologie, to trovano the mutazione genetica che è responsabile per the segni e sintomi espressa come parte del malattia mitocondriale. The ‘genotipo’ si riferisce nel specifico genetica anormalità che causa the malattia. Nel caso di una malattia mitocondriale, questo gene mutazione può avvenire either in the DNA mitocondriale o in the DNA nucleare. Over the last due decadi, the maggiore sforzo è stata to identificare the mutazioni sulla DNA mitocondriale che sono responsabile per the malattia. Comunque, è becoming increasingly evidente che nucleare gene mutazioni deve anche be responsabile per alcune, e possibilmente La maggioranza, of tutti malattie mitocondriali. The ‘fenotipo’ si riferisce to how the persona expresses l'anormalità genetica, specificamente the segni e sintomi del malattia. E' qualcosa unica to malattie mitocondriali che individual membri of a famiglie con l'esatto gene mutazione, o genotipo, può avere differenti clinica expressions, o fenotipi. Viceversa, persone con the similare manifestazioni cliniche può avere una varietà of differenti gene mutazioni. Advances in genetica molecolare e medicina mitocondriale occurring in the 1980s e 1990s resulted in identifying a numero del DNA mitocondriale mutazione puntiforme (the genotipo) specificamente associati con the sindrome che historically è riferite to come MELAS (the fenotipo). E' importante ricordare che the acronym ‘MELAS’ è usati descrivere those con citopatie mitocondriali che hanno acidosi lattica e ictus o simil-ictus eventi come parte of loro clinica espressione. Sebbene alcune mutazione puntiforme per MELAS sono state identificata e sono facile to esame per, non tutti i pazienti con clinica MELAS harbored the conosciuta MELAS mutazione puntiforme. |
UMDF 52 Think Mitocondri
| Ictus Ictus sono comune neurologici eventi che risultato in permanente cervello danno e sono solitamente causato by a mancanza di ossigeno o sangue flow to a regione del cervello. Ictus sono ora spesso riferite to come “cervello attacchi”, come uno dovrebbe refer to cuore attacchi. Outside the setting di una malattia mitocondriale, the blockage avvengono come parte del processo of atherosclerosis, quando a waxy sostanza builds up sulla inside del arteries e finally causa the complete blockage of che artery. In altro circostanze, a sangue clot o altro sostanza can travel attraverso the corrente sanguigna e blocco the artery, con è chiamati an embolus. Questo interruption in sangue flow o ossigeno causa the affetto area del cervello di essere deprived dell'energia. Come the energia immagazzinati sono depleto the affetto cervello cellule die. The estesa del danno è determinata by il quantitativo of cervello coinvolti. Ictus può anche be causato by bleeding into il cervello, la quale può avvenire se a persona ha molto alto pressione sanguigna o a cervello tumore. When bleeding avvengono, tessuto cerebrale è injured e causa perdita of neurologici funzione. Modern technologie permette il dottore per confermare the clinica sospetto of a ictus by prestazioni an MRI scansione del cervello. Sintomi of a ictus possono includere perdita di visione, debolezza, sensoria loss e intellettuali problemi. Sebbene a ictus defines a permanente danno, i sintomi può migliorare nel corso del tempo. Il processo of neurologici recovery non è ben understood, ed non è ben conosciuta perché alcune pazienti migliora e altri do non. In alcune casi, the neurologici event è quickly reversibile, con the debolezza o perdita di visione disappearing in minuti to ore, e questo event è comunemente riferite to come una TIA o transitori ischemica attack. Uno comune cause of TIAs sono pensarono di essere dovuta al blockages in sangue flow che quickly reverse stessi. We tend to think che ictus principalmente avvengono in older persone, ma they può avvenire a ogni età. Ictus sono state descritte in the precoce rapporti dei pazienti con malattie mitocondriali, e we will explore those associati con citopatie mitocondriali. Ictus che avvengono in the setting di una malattia mitocondriale sono solitamente non dovuta al ogni del fattori elencato sopra, ma sono più probabilmente dovuta al a carenza della produzione di energia in una particolare regione del cervello. Cervello cellule, like altro cellule, |
necessitano energia to funzione e survive. Questo energia è delivered nel sangue stream come glucosio (o altro sostanze like chetoni). In pazienti con malattie della fosforilazione ossidativa, the sangue carrying the glucosio, chetoni e ossigeno è delivered appropriatamente, ma la capacità di produrre energia in la forma di ATP è diminuita. Questo mancanza di energia può provocare in cervello danno, just come se the sangue carrying the glucosio e ossigeno mai got to il cervello perché of a blockage in the artery. Riassumendo, the ictus è dovuta al a critiche energia crisi, e the estesa del energia crisi will determine the size e gravità del ictus. In alcune pazienti con malattie del metabolismo energetico, ci può essere a sistemiche incapacità per generare proper amounts of either glucosio o corpi chetonici, solitamente in the setting of relative inedia o affezioni; questo mancanza di energia substrate può anche risultato in a ictus. Come con più comune tipi of ictus, those con malattie mitocondriali può avere breve eventi, con lievi o grave sintomi che quickly reverse. Qualche volta transitori neurologici eventi sono accompagnata by a cefalea. These cefalee sono similare nel comune emicranie cefalee che affect milionis delle persone. The cefalee tend to coinvolgono solamente uno side del capo a a time. i pazienti solitamente describe il dolore come “pounding.” In pazienti con malattie mitocondriali, transitori neurologici eventi con o senza cefalee può avvenire e può essere visionate come una reversibile “simil-ictus eventi.” The gradation fra the transitori neurologici sintomi associati con complicata cefalee, reversibile simil-ictus eventi, e ictus è probabilmente continua, con the irreversible ictus representing the più grave end dello spettro. Sintomi of a ictus o simil-ictus event can comprendono: • Various modelli of visual disturbazione o perdita di visione • Debolezza of vario gradi, generalmente colpente solamente uno side del corpo • Clumsiness, generalmente colpente solamente uno side del corpo • Incapacità to speak appropriatamente o understand parola • Mental confusione, loss of perceptual capacità, o in grave casi, loss of consciousness |
Think Mitocondri 53 UMDF
Tabella 1: Problems That Maggio Be Associated con Mitocondriale Citopatie
Organ System Possible Problems
Cervello ritardo nello sviluppo, ritardo mentale, demenza, attacchi epilettici,
neuropsichiatrici
disturbi, atipiche paralisi cerebrale, cefalee, ictus
Nervi debolezza (le quali possono essere intermittente), neuropathic dolore, assenti riflessi,
gastrointestinale problema (ge riflusso, costipazione, pseudo-ostruzione),
fainting, assenti o eccessivo sudorazione derivante in temperature regolazione
problemi
Muscoli debolezza, ipotonia, cramping, dolore muscolare
Reni prossimale renale tubulare deterioramento derivante in loss di proteine, magnesium,
phosphorous, calcio e altro elettroliti
Cuore cardiaca difetti di conduzione (blocco cardiaco), cardiomiopatia
Fegato ipoglicemia (poco zucchero nel sangue), insufficienza epatica
Occhi perdita di visione e cecità
Orecchie perdita di udito e sordità
Pancreas e Altro
Glands
diabete e esocrine insufficienza pancreatica (incapacità a fare gli enzimi digestivi), paratiroide insufficienza (calcio basso)
Sistemico insufficienza to gain peso, short statue, affaticabilità, problemi respiratori
comprendenti intermittente air hunger, vomito
| Dato the grande fabbisogni di energia del cervello, è no wonder perché ictus could avvengono in those con OXPHOS malattie. Comunque, ci sono still più questioni than answers con respect to ictus. It non è pienamente understood: • What can trigger a ictus dopo anni of normale funzionamento • Why ictus tend to coinvolgono piuttosto specifico parti del cervello che non sono solitamente affetto outside the setting of citopatie mitocondriali • Why ictus avvengono in alcune pazienti con altrimenti minimal o no evidenze of prima cervello coinvolgimento, mentre altro pazienti con grave epilessia e ritardo mentale mai Hanno a ictus • Why un paziente con a grave compromissione of OXPHOS funzione come misurato da muscoli analisi dovrebbe mai Hanno a ictus, mentre un paziente con a lievi compromissione may Hanno a ictus Melas Come mentioned earlier, the più ben-descritte sindrome nelle quali ictus sono conosciuta to avvengono è MELAS, o Mitocondriale miopatia, Encefalopatia, acidosi lattica e Ictus-Like episodi . The sindrome della MELAS was proposti in 1984, e was usati descrivere pazienti con normale precoce sviluppo, crescita ritardare, attacchi epilettici, acidosi lattica, sfilacciate-rossi fibre on biopsia muscolare (signifying overproduction of anormale mitocondri) e episodi che resembled ictus. The |
ictus do non sempre follow the modello che sono visto in ictus dovuta al blocked sangue vessels. Il paziente can pienamente recover from the ictus o il paziente possono svilupparsi a progressive neurologici sindrome coerente di di molteplici ictus con o senza attacchi epilettici. Alcuni pazienti con MELAS may anche Hanno altro problemi neurologici che sono dovuta al an insufficienti energia supply nel cervello e altrove nel corpo. Questo comprende atassia (clumsy movimenti), atrofia ottica e retinopatia (con o senza perdita di visione), distonia/miocloni/corea (anormale twisting o shaking movimenti), e neuropatia (nerve danno). Neuro-psichiatrici sindromi può essere a transitori o progressive problema in MELAS e altro mitocondriale cytopthies. The ictus in MELAS solitamente avvengono fra the ages of 5 to 15 anni, ma può avvenire a ogni time. The ictus generalmente coinvolgono posterior regioni del cervello, specialmente the occipitale lobes (colpente vision) o posterior parietal o temporal lobes. The iniziale ictus in MELAS può essere grave, o il paziente may Hanno a emicranio-simili event quando il paziente develops debolezza, perdita di visione, o mental cambiamenti associati con a sever cefalea. The neurologici event may reverse (come sarebbe con a emicranie) o may non. All pazienti con MELAS Hanno similare sintomi, ma like tutti malattie mitocondriali, la gravità dei sintomi vary from paziente to paziente, persino in quei pazienti che sono membri of lo stesso famiglie. Questo variabilità è dovuta al a numero of |
UMDF 54 Think Mitocondri
| fattori. E' critiche to note che the sindrome della MELAS non è dovuta al una singola mutazione genetica. About 80% dei pazienti con MELAS Hanno an A to G point mutazione a base pair 3243, con altro pazienti avendo a T to C point mutazione a bp 3271 o a mutazione of G to A a bp 3252. Tutti questi mutazioni affect the transfer RNA che codifica per isoleucina (isoleucina UUR tRNA). Secondo , come con tutti altro citopatie mitocondriali dovuta al a mutazione nel mtDNA, the percentuale of anormale mtDNA varia randomly fra the differenti tessuti nel corpo. In altro words, il cervello of uno paziente may Hanno più anormale mtDNA than i muscoli of che stessa paziente; conseguentemente the ictus manifestazioni può essere più o meno evidente than the altro caratteristiche del condizione. Infine, pazienti con lo stesso mutazione in lo stesso famiglie may inherit variabile quantità of anormale mtDNA, con significa che il cervello of uno famiglie membro può essere più anormale mtDNA, e di conseguenza più grave neurologici malattia, than il cervello di un'altra famiglie membro. Ictus Outside the Setting of Melas It non è infrequente per a persona con citopatia mitocondriale to Hanno molti of lo stesso caratteristiche della MELAS, comprendenti the miopatia, encefalopatia, acidosi lattica e ictus, ma esame negative per la mutazione puntiforme associati con MELAS. Come we learn più circa la mutazione puntiforme, biochimica, catena respiratoria associati strutture e ultra-structure del mitocondri, we may understand più circa what causa ictus. Prevenzione, La diagnosi e Trattamento Non ci sono provato preventative misure che will reduce il rischio of ictus in pazienti con malattie mitocondriali. Inoltre, in pazienti con MELAS, there probabilmente è no trattamento today che will prevenire a ictus. Illnesses tali come the flu può aumentare il rischio of a ictus o simil-ictus event, ma la maggior parte delle ictus avvengono outside the setting di tali an affezioni. Ictus-like eventi e neurologici deteriorazione sono state descritte dopo infezione virale (con susseguente disidratazione e temporary inedia), vaccinazioni, alcool ingestione, hypoxia (come dovrebbe avvengono a molto alto altitudes o con cigarette il fumo), ingestione of |
monosodium glutamato (MSG) e esposizione to estrema temperatures. E' impossibile to eliminate esposizione to viruses, e come una generale rule, bambini con citopatie mitocondriali should Hanno routine vaccinazioni. E' a materia of personal scelta to evitare alcool, cigarette smoke e MSG. Aside from locations tali come San Francisco, it non è possibili to evitare temperature extremes, e se a persona mostrano una particolare sensibilità to estrema caldo o freddo, these devono essere evitati. Senza riguardo, proper bundling è importante in the winter, e over-dressing può essere come pericoloso come sotto-dressing. In the summer, overheating può essere Particolarmente dannoso nel bambino da una malattia mitocondriale, così loose fitting clothes, proper shading (a cool hat), plenty of acqua, e avoidance of prolungato diretta sunlight è importante. Senza riguardo, stretta adherence to tutti del sopra will non prevenire a ictus from occurring, nor è there ogni evidenze che uno can minore il rischio of ictus in the setting della MELAS, persino con avoidance of tutti precipitating fattori e use of medicamento e vitamine. The valutazione of a persona con a ictus del first simil-ictus event dovrebbe comprendono a esaminazione fisica e neuroimaging, preferibilmente con imaging a risonanza magnetica (MRI). In the situazione del first event quando the persona ha yet di essere diagnosticata con a citopatia mitocondriale, the MRI can aid in la diagnosi se the modello of cervello danno fits con the tipico modelli visto in queste malattie. Once the clinica modelli of simil-ictus eventi è stata stabiliti, ripetuta imaging non è necessarie con ogni event. Non ci sono specifico trattamento per ictus che avvengono in persone con una malattia mitocondriale. The nuovi trattamenti per ictus che avvengono outside the setting delle malattie mitocondriali, tali come tPA, non sono beneficial per the ictus che sono dovuta al disfunzione mitocondriale. Supportive care è l'essenziale e should consist of bed riposo, e in grave casi intravenoso liquidi può essere necessaria. Metaboliche parametri tali come elettroliti, glucosio, lattato e ammoniaca devono essere esaminati immediatamente e come necessaria durante recovery. Low glucosio ematico devono essere trattati immediatamente. Se il paziente non è alert abbastanza to eat, nutrizionale supporto è necessarie. |
Think Mitocondri 55 UMDF
| Conclusion Aside from MELAS, nelle quali, by definizione, ictus è parte del sindrome, it non è possibili per determinare il rischio of ictus in a persona con citopatia mitocondriale. Persino in those con MELAS, the timing del ictus e simil-ictus eventi non sono predictable e avvengono a random. It sembri di essere the sperimentano of those che care per a lot dei pazienti, che esaminazioni positive per the conosciuta mutazione puntiforme associati con MELAS generalmente carries con it a worse overall prognosi, in termini of capacità per recuperare from the ictus. Se o non la maggior parte delle persone con malattie mitocondriali sono a |
aumentato rischio of ictus over loro lifetime non è conosciuta, sebbene meno grave simil-ictus eventi sono conosciuta to avvengono piuttosto frequentemente in alcune. Disorders of le funzioni cerebrali che avvengono in persone con citopatie mitocondriali possono includere attacchi epilettici, ritardo nello sviluppo e grave cognitive problemi che comprendono ritardo mentale e autismo, demenza (loss of acquisite cognitive funzioni), stokes e simil-ictus eventi. Again, non tutti persone con citopatie mitocondriali Hanno malattie of le funzioni cerebrali, e those che do may Hanno lievi, moderate o grave sintomi. |
UMDF 56 Think Mitocondri
Prenatal La diagnosi of Mitocondriale malattia
by Brian H. Robinson, Ph.D.
Hospital per Sick bambini/University of Toronto
© The Mitocondriale News, United Malattie mitocondriali Foundation, 1999
| Introduction The malattie del metabolismo energetico comprise un gruppo of talvolta devastante malattie. They solitamente Hanno loro insorgenza in infanzia e può essere fatali dovuta al loro rapid progressione in a materia of settimane, mesi o anni. A più piccola subset dei pazienti Hanno slower sviluppare malattie con insorgenza in the teenage o adulto anni, ma può essere a rischio per avendo un bambino who può essere molto più gravemente affetto. How do we deal con questo from the point of view of prenatal diagnosi? How do we counsel genitori circa the chances of avendo un'altra affetto bambino, e how do we counsel them circa the prenatal rilevazione del malattie mitocondriali come una gruppo? A search of la letteratura will non trovano molto molto scritto circa ogni di queste questioni. The mancanza di letteratura deriva from due maggiore problemi. La prima problema è che molto piccolo è conosciuta circa the modello of inheritability di molti di queste malattie. In alcune we assume autosomica recessiva the mode dell'ereditarietà by default. In altri we just do non know whether the difetto è nucleare o mitochondrially codificato in ogni uno famiglie. The second problema è the mancanza di a collective database di informazioni per those medici, scienziati e genitori brave abbastanza to go attraverso prenatal diagnosi procedure con no guaranteed risultato. Ogni genitori reading questo, who poi ask the question “è prenatal diagnosi disponibili per us?” necessitano to Hanno a loro disposal come molto informazioni come possibili circa il disturbo che ha affetto loro famiglie. Le maggiori informazioni you necessitano è: 1) What è the name attached to questo disturbo? 2) Is there an enzimi diagnostici esame per questo disturbo? 3) Has il disturbo been narrowed down to il livello del gene? 4) Can il disturbo be rilevata in skin fibrolasts? |
Why These Domande? The name attached nel disturbo tells you either circa the symptomatology del processo di malattia, o circa the name del principle enzimi che (by interrupting biochimica percorsi) è responsabile per the symptomatology del processo di malattia. It può essere an acronym tali come MELAS, MERRF o NARP, o it può essere a name like citocromo ossidasi carenza o PDH complesso carenza. Questo informazioni in alcune casi è abbastanza per you discernere whether prenatal diagnosi è possibili. Very spesso you anche necessitano sapere the subtype del disturbo. Your medico possono aiutare you con questo se you don’t know the rispondere nel question destra ora. E' al di fuori dello scopo di questo articolo to fill in the differenziale diagnostici caratteristiche of tutti malattie mitocondriali così in the table sotto Ho set out ogni del malattie con conosciuta subtypes e alcune informazioni con potrebbero aiuto circa prenatal diagnosi. What Are You Probabilmente To Find? La semplice truth a the moment è che the minority di queste malattie è passibili to prenatal diagnosi. That è the bad news. The buon news è che the situazione è il miglioramento tutti the time. The miglioramento comes from due fonti, la prima being miglioramenti in cellulari rilevazione tecniche. Better esaminazioni procedure sono being sviluppato tutti the time in laboratori attorno the mondo. The second fonte of discovery è the descrizione del geni e genetica mutazioni responsabile per questo gruppo di malattie. Questo anno the gene difetto per PDH-X carenza, uno forma of Complesso I carenza (AQDQ gene difetto), e uno forma of La carenza di COX (classica malattia di Leigh dovuta al the SURF1 gene) sono state descritte. Più sono sulla way. (The a seguito più dettagliata informazioni è divided into discussione of DNA nucleare difetti e mtDNA codificato difetti.) |
Think Mitocondri 57 UMDF
| Nucleare-Encoded I difetti: Ci sono a lot di queste e la maggior parte delle sono ereditata in an autosomica recessiva modello. That significa che sebbene the genitori sono unaffected they entrambi contribuiscono a mutazione in loro geni nel affetto bambino. Piruvato Dehydrogenase Deficiency: La maggior parte dei pazienti avendo questo difetto Hanno a difetto in loro E1a gene con è carried sulla X-chromosome. Perciò the affetto subtype è E1. Comunque, questo è an insolita collegata a X malattia perché entrambi boys e girls può avere it. When it avvengono, la maggior parte delle del time the difettosa gene non può essere rilevata in the madre o the father. Questo è perché la mutazione happened in la formazione del egg o sperm ma non è presenti in the altro tessuti del genitore. Questo è buon news per those seeking prenatal diagnosi perché il rischio di ricorrenza drops from 1 in 2 per boys to a molto basso numero. To my knowledge, 32 prenatal diagnosi sono state fatto per questo difetto, e 30 sono state unaffected, abbastanza normale bambini. Da quando genetica molecolare diagnosi per questo malattia è feasible, questo è the metodi of scelta. Measurement delle attività enzimatica può essere risky, specialmente se the expected bambino è female. The altro difetti - E2, E3 e X ñ sono tutti autosomica recessiva e potrebbero rilevata either by molecolare tecniche o by misurazione delle attività enzimatica. Piruvato Carboxylase Deficiency: Questo difetto è stata rilevata prenatally by enzimi misurazione. Molecular tecniche may supercede questo, ma sono molto più costosi. Respiratory Chain I difetti Complesso I (NADH-Ubiquinone Oxidoreductase): Questo è a molto difficile enzimi per misurare in colture cellulari, così cercando per rivelare it in amniocytes o chorionic villous cellule è next to impossibile. Comunque, the più grave difetti in questo enzimi può essere rilevata by the alto lattato a piruvato rapporto presenti nelle cellule. Eighteen prenatal esami per questo difetto sono state fatto by questo tecnica. Fifteen were predicted unaffected, tre were affetto. Uno di queste previsioni was incorretta by questo metodo. Better metodi per questo difetto will come come più molecolare difetti sono definita. |
Complesso II (Succinato-CoQ Oxidoreductase): I difetti in the subunità of succinato deidrogenasi sono state definita in molecolare termini. Questo devono essere passibili to prenatal diagnosi in the futuro. Complesso III (Nucleare): No Complesso III difetto è stata definita in molecolare termini. Questi difetti sono poorly espressa in fibroblasti, e di conseguenza probabilmente non passibili to prenatal diagnosi in amniocytes. Complesso IV (Citocromo Ossidasi Deficiency): The gene per the più comune forma of citocromo ossidasi carenza ha recentemente been shown by Dr. Eric Shoubridge di essere the SURF1 gene, a gene responsabile in alcune way per the assemblaggio del citocromo ossidasi complesso. Questo difetto was already diagnosable in amniocytes by enzimi tecniche. The meno comune Quebec- Saguenay Lac St Jean La carenza di COX è causato by a differenti gene localizzate on Chromosome 2 (BHR). E' difficile per diagnosticare prenatally, ma un nuovo tecnica basato on Chromosome 2 marcatori è being sviluppato. The altro forme of La carenza di COX non sono ben espressa in fibroblasti, e piccolo informazioni on prenatal diagnosi è stata pubblicati. mtDNA Depletion sindrome: Mentre questo sindrome entails gradual loss del mtDNA, the gene responsabile è a nucleare uno e probabilmente è responsabile per alcune critiche funzione in mitocondriale mantenimento. The difetto è solamente talvolta espressa in cultured cellula sistemi, con significa che osservando a amniocytes o chorionic villous cellule può non essere affidabile. Le maggiori enzimi deficit è citocromo ossidasi carenza e the malattia è talvolta mistaken per a primaria difetto in questo enzimi. Prenatal diagnosi ha non been attempted in questo malattia to my knowledge. mtDNA Deletion sindrome: In questo difetto, delezioni multiple del mtDNA appare in tessuti, solitamente muscoli scheletrici e cuore. La malattia è spesso autosomico dominante in ereditarietà, con significa you can inherit the malattia from uno genitore ed è di nuovo a codificate dal nucleo gene con è responsabile. Numerose chromosomal |
UMDF 58 Think Mitocondri
| loci sono state localizzate in differenti famiglie, e prenatal diagnosi might be possibili ma per quale è spesso an adulto insorgenza malattia, c'è understandably non a lot of interest in questo procedure. Deletions non sono spesso visto in colture cellulari. |
mtDNA Deletion (sporadica, Kearn Sayre sindrome, CPEO, sindrome di Pearson): Solitamente the delezioni non sono visto in colture cellulari e the affetto casi sono sporadica, cioè non collegata to altro famiglie membri, e raramente presenti |
Expressed in
Defect Ereditarietà
Mode
Molecular
Test
Available
Muscle I fibroblasti Amniocytes CVS Prenatal Test disponibili
PDH
E1a
E2
E3
X
X-L; AD; AR
AR
AR
AR
Yes
Yes
Yes
Yes
Yes*
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Activity (Males)
Molecular (Femmine)
-
-
Yes
PC Nucleare AR Yes X Yes Yes ? Yes
Complesso I Nucleare AR X Yes Yes ? Under L'indagine
Complesso II SDH Nucleare AR Yes
(alcune)
Yes ? ? ? ?
Complesso III AR Yes Yes ? ? ? ?
Complesso IV Nucleare
Classic malattia di Leigh
SLSJ malattia di Leigh
Fatal Infantile
Cardiomiopatia
AR
AR
AR
AR
Yes
X
X
X
Yes
X
Yes
Yes
Yes
X
Yes
Yes
X?
Yes
?
?
?
Enzyme Le analisi
Under L'indagine
??
mtDNA Depletion sindrome
Nucleare
AR X ? ? ?
mtDNA Deletion (Familial)
Nucleare
AR; AD X Yes X ? ? ?
mtDNA Deletion (Sporadica) S o M Yes Yes X ? ? ?
mtDNA tRNA (MELAS,
MERRF)
M Yes Yes ? ? ?
mtDNA rRNA M Yes Yes Yes ? ? ?
mtDNA Protein (NARP) M Yes Yes Yes ? ? Under L'indagine
Legend AR = autosomica recessiva AD = autosomico dominante S = sporadica M =
materna (attraverso mtDNA) X-L = collegata a X
How to Read the Tabella
Prima di tutto, locate the name del difetto, poi look per the subtype. Per
instance, se you know your bambino ha
PDH complesso carenza, the informazioni è differenti dipendendo on whether the
difetto è localizzate in the E1, E2,
o X componenti. Se you sono osservando up citocromo ossidasi carenza you necessitano sapere se you sono dealing
con the classica Leigh forma, the Saguenay Lac St Jean Leigh forma, the fatali
infantile forma, the cardiomyopathic
forma, o a non-classificate varietà. Per ogni of them the informazioni è
differenti.
Under ogni subtype è elencato:
Nucleare/Mitocondriale: Is questo disturbo determinata by the DNA nucleare o by
DNA mitocondriale (mtDNA)?
Molecular: Questo says whether the gene responsabile è stata identificata, si o
no.
Muscle, I fibroblasti: These headings tell you whether the enzimi difetto è
measurable in fibroblasti (cultured
skin cellule).
Amniocytes: Questo tells you whether fetal cellule in the amniotic liquidi express the
difetto
CVS: Questo tells you se è conosciuta whether chorionic villous cellule from the
placenta express the difetto
Prenatal La diagnosi: Questo tells you whether prenatal diagnosi per questo disturbo
è stata condotte con pieno successo.
Think Mitocondri 59 UMDF
| in fratelli e sorelle. Questo puts the chances of avendo due affetto bambini a a basso probabilità. E' doubtful che osservando per deleted mtDNA in amniocytes o chorionic villous tessuto sarebbe utili. La principale DNA mitocondriale Disorders These mtDNA difetti sono ereditata maternamente. Per those mothers che the gene difetto, c'è piccolo di essere gained by esaminazioni the fetus, come the fetus will anche Hanno the gene difetto, e perché the percentuale of anormale mtDNA in the tested cellule may non riflette the percentuale of anormale mtDNA in i muscoli o cervello, non c'è metodi predire how sick the baby sarebbe. C'è stata piccolo interest in prenatal diagnosi per those mtDNA difetti che coinvolgono transfer RNA geni. Questo comprende the la maggior parte delle comune malattie, 3243 MELAS e 8344 MERRF. The amount of mutated mtDNA in ogni uno tessuto determina whether il tessuto è affetto. The amount o estesa of mutated mtDNA in muscoli, cuore e cervello of a sviluppare fetus may bear no relationship a tutti to il quantitativo che può essere misurato in amniocytes o chorionic villous tessuto. Questo ha precluded ogni attempt per diagnosticare these difetti in utero. Per numerosi malattie colpente proteine coding geni del mtDNA, specialmente 8993 NARP, the situazione può essere differenti. Mentre sperimentano è limitato it sembri che amniocyte livelli of la mutazione può riflettere a meno partially the livelli in the tessuti del fetus. Obviously più dati è necessaria in questa area, ma a meglio the predictability di tali esaminazioni è solamente probabilmente per fornire an approximate guide nel percentuale of la mutazione. Per |
altro condizioni tali come the retinopatia pigmentosa associati con the11778 LHON mutazione, no informazioni è disponibili, ma di nuovo, per ovvia ragioni c'è piccolo interest in prenatal determinazione of status. Facilities Per Prenatal La diagnosi Having read the sopra guide, you può essere interested in pursuing the soggetto ulteriori. The uno course of action che non è opportuno è to begin a gravidanza sulla basis of what you know, e poi seek out prenatal diagnosi. The due maggiore problemi che ogni centri embarking on a prenatal diagnosi Hanno to deal con sono: 1) Being satisfied che the criteri necessaria per prenatal diagnosi cioè: espressione del difetto in colture cellulari, sono state met. 2) Fear of litigation, should a prediction be incorretta. La prima problema è uno che spesso needs di essere looked a molto attentamente e stessa may take numerosi mesi per risolvere. The second problema è più difficile e prevents molti institutions from attempting prenatal diagnosi per malattie mitocondriali. In either caso, lengthy discussioni del facts surrounding your caso e a realistic exploration of expectations con your genetica counselor o medico sono obbligatorio prima you do qualcosa. Il riferimento Poulton, J. e Marchington, D.R. (1996) Prospects per basate sul DNA prenatal diagnosi delle malattie mitocondriali. Prenatal La diagnosi 16:1247-1256. |
continuati on page 60
UMDF 60 Think Mitocondri
Vomito coclico in Mitocondriale malattia
by Richard G. Boles, M.D., Division of Medical genetica, Childrens Hospital Los
Angeles
© The Mitocondriale News, United Malattie mitocondriali Foundation, 2001
| Gastrointestinal (GI) sintomi sono comunemente incontrati in pazienti con una malattia mitocondriale. Più spesso, sintomi sono episodico in che they come e go, e sono relativo to ‘funzionale’ problemi del intestino. In particolare, vomito è comune fra sufferers di molti differenti malattie mitocondriali, e fra these persone, vomito stessa ha molti differenti causa. Occasional bouts of vomito è comune, specialmente in infanti, e è spesso trovati di essere causato by riflusso gastroesofageo . Questo articolo discusses uno particolare tipo of vomito disturbo, chiamati ‘cyclic vomito’. Vomito coclico non è nuovi come it was first descritte in the eighteenth century, sebbene persino today molto poche medici o altro clinica care providers Hanno heard of it. Vomito coclico si riferisce to discrete e grave episodi of vomito, nausea e lethargy (grave tiredness). vomito fra episodi . preferring to lie down in a dark e/o quiet place, e non interested in ogni del attività della vita. vomito e perdita di appetito può essere grave abbastanza to necessitate ospedalizzazione per intravenoso (IV) liquidi con ogni episodio. In altro casi, nausea e lethargy può essere molto più troublesome than vomito. triggered by fisica o psicologico sforzo, o appare to come a random. Ciascuno episodio can last per ore a molti giorni, ma solitamente c'è a caratteristiche durata in ogni individual paziente. In alcune casi, an episodio può essere stopped se the bambino sleeps. Alcuni sufferers Hanno ulteriori sintomi durante episodi tali come loose stools, drooling o cefalea. Ci possono o non può essere an ‘aura’, o sintomi con avvengono prima vomito begins. Nella maggioranza dei casi, cyclic vomito iniziano in bambini ranging from circa età 3 to 8 anni, sebbene il disturbo can partenza a ogni età comprendenti in infanti e adulti. Vomito coclico can run its course e resolve, continue indefrinitamente, o cambiano into |
emicranie cefalee. La maggior parte sufferers Hanno normale intelligence e sono generalmente sano fra episodi , comunque, molti of them Hanno vari gradi of ritardo nello sviluppo e/o ulteriori problemi tali come epilessia. Vomito coclico ha molti conosciuta causa, comprendenti intestinale blockage, cervello malattie, reni malattia, e numerosi differenti metaboliche malattie. Molti di queste causa sono trattabili, e a attenta diagnostici lavoro-up è importante. Comunque, in the vast majority di casi, none del sopra causa può essere trovati, e these persone sono dati la diagnosi of ‘cyclic vomito sindrome’ o ‘CVS’. Cefalea emicranica e episodico grave dolore addominale (addominale emicranie) sono molto comune in CVS sufferers e loro famiglie membri alike (solitamente in the materna parenti!). At presenti, emicranie cefalee, addominale emicranie e CVS sono considerate di essere relativo, e possibilmente sono differenti manifestazioni of lo stesso disturbo. Vomito coclico è stata riportati in persone con the A3243G ‘MELAS’ mutazione in the DNA mitocondriale (mtDNA). In aggiunta, uno bambino con CVS, ritardo nello sviluppo, attacchi epilettici, crescita ritardare e ulteriori problemi was riportati to Hanno a grande delezione nel mtDNA e duplicazione. Comunque, in my sperimentano come una metaboliche geneticist, CVS con o senza ulteriori problemi non è rara in bambini con malattie mitocondriali, e fra questo gruppo, ‘routine’ l'analisi del mtDNA fails to identificare previously conosciuta mutazioni del mtDNA nella maggioranza dei of them. To date, Ho personally esaminati circa 15 bambini con CVS e sospetta malattia mitocondriale. These e an ulteriori 50 casi raccolti mondowide con CVS e ulteriori neuromuscolare problemi (un gruppo a rischio per possibili malattia mitocondriale) sono state entered into an con l'andare del tempo ricerche studio. Molti di queste bambini Hanno a specifico modello di ulteriori clinica e laboratorio rilevamenti comprendenti GI dismotilità (riflusso, delayed gastric emptying, costipazione), disautonomia (inspiegata fevers, alto ritmo cardiaco, ecc.), debolezza muscolare, cronica affaticabilità, attacchi epilettici e dolore (capo, |
Think Mitocondri 61 UMDF
| abdomen e/o estremità). The latter, riferite to talvolta come “muscoli cramps”, è occasionalmente associati con swelling e skin discoloration in a manner indicativa of neuro-vascolare distrofia. No singola bambino suffers from tutti di queste problemi, e quando presenti in un dato bambino i sintomi tend di essere episodico e variabile. In alcune di queste bambini, cyclic vomito stessa è a minor parte del bambino’s problemi, e may disappear o mai sono state presenti. Intelligence ranges from gifted to grave ritardo mentale. Laboratory analisi in bambini con CVS e malattia mitocondriale dimostra elevati acido lattico e anormale gli acidi organici urinari (chetoni, Kreb ciclo intermedi, e/o ethylmalonate) precoce in episodi di vomito , ma biochimica esami sono raramente anormale a altro times. Alcuni bambini Hanno ricevuto muscoli biopsie con revealed rilevamenti indicativa di disfunzione mitocondriale, comprendenti aumentato variazioni in dimensioni delle fibre, proliferazione mitocondriale , e/o complesso 1 carenza. Comunque, in my opinion, the la maggior parte delle striking rilevamenti è ereditarietà materna of lo stesso kind of episodico problemi visto in the affetto bambini stessi, ma solitamente to a lesser grado, comprendenti emicranie, cyclic vomito, GI dismotilità, disautonomia, debolezza muscolare o dolore, cronica affaticabilità, e/o attacchi epilettici. At the time di queste writing, a meno 5 unrelated casi were trovati to Hanno eteroplasmico (due differenti mtDNA sequences presenti in lo stesso individual) nucleotide cambiamenti nel mtDNA control regione. These molecolare variants sono ereditata maternamente (presenti in madre e fratelli e sorelle, persino se they stessi sono senza sintomi) e were non trovati in over 100 bambini senza malattia mitocondriale. La stessa control regione variants were trovati in bambini con una malattia mitocondriale ma senza CVS, e la significatività of our recente rilevamenti non sono yet chiara e sono the soggetto of con l'andare del tempo investigazione. Comunque, our dati does dimostrano che malattia mitocondriale con cyclic vomito è spesso ereditata maternamente . Improbabili la maggior parte delle pubblicati casi con malattie mitocondriali, malattia progressione appears di essere rare in bambini con maternamente-ereditata cyclic vomito. Uno eccezioni nel generale benigna malattia course è che a few famiglie Hanno had infanti sotto età 2 anni who died suddenly e were labeled come |
“SIDS”. La maggior parte bambini, e specialmente loro affetto parenti, attend normale schools o Hanno jobs/careers, e loro lives sono abbastanza normale fra malattia episodi . In molti scuola-aged affetto bambini, grave affaticabilità e debolezza muscolare ha necessitated the occasionale usage of wheelchairs e/o metà al giorno o home schooling. All troppo spesso, clinica care providers e/o scuola personnel Hanno down-played il processo di malattia, persino nel estesa of labeling the bambino/famiglie come exaggerating sintomi, being psychologically disturbed, o avendo causato the affezioni (Münchausen By Proxy). The buon news è che trattamenti sono disponibili per cyclic vomito in persone con una malattia mitocondriale. In malattia mitocondriale, sintomi sono believed to avvengono quando energia supply cannot meet domanda di energia. Da quando spesso piccolo può essere fatto per aumentare energia supply, decreasing domanda di energia è a maggiore parte della terapia. In pratici termini, questo significa the riduzione of sforzo, comprendenti the avoidance of digiuno, limitando esposizione to ambientale temperature extremes, e the prompt trattamento of infezioni e disidratazione. Vomito coclico e alcune altro sintomi spesso migliora con frequente feedings of complesso carboidrati, comprendenti fra pasti e a bedtime. Altro bambini migliora se awakened durante sonno per a spuntino e/o collocate on a basso grassi dieta. In aggiunta to fisica sforzo, the riduzione of psicologico sforzo è importante: non perché questo è the cause del malattia, ma perché sforzo aumento domanda di energia e can trigger an episodio. In casi nelle quali the risposte to these semplice misure non è adeguate, anti-emicranie medicamento comprendenti amitriptilina (Elavil), ciproeptadina (Periactin) o propranolo (Inderal) taken a bedtime o più spesso can reduce il numero of episodi di vomito nella maggioranza dei casi, talvolta dramatically. When they do avvengono, episodi di vomito sono trattati con liquidi endovena (10% Dextrose con standard elettroliti a a rate of 1.5 to 2 times mantenimento) in a dark e quiet room in order facilitare sonno. In alcune casi, ondansetron (Zofran) e/o farmaci to induce sonno (cioè lorazepam/Ativan) sono aiuto. Diagnostic lavoro-up (esaminazioni) devono essere tailor-fit to ogni individual bambino. Naturalmente, confirming la diagnosi di malattia mitocondriale e ruling out altro trattabili metaboliche malattie (ciclo dell'urea dis |
UMDF 62 Think Mitocondri
| orders, acidemie organiche) devono essere pursued. I suggerire che a minimum lavoro-up devono includere siero elettroliti, routine urinalysis, lattato plasmatico, quantitative aminoacidi plasmatici e quantitative gli acidi organici urinari (comprendenti full quantificazione of Kreb ciclo intermedi e altro potenziale ‘mitocondriale marcatori’), con campioni ottenuto precoce in a grave o tipico vomito episodio. DNA mitocondriale analisi devono includere a a minimum PCR per A3243G e Southern blotting. Unless la diagnosi di malattia mitocondriale è rigidi e CVS sintomi rispondono to trattamento, lavoro-up per altro potenziale causa of cyclic vomito devono essere condotte, possibilmente comprendenti ma non necessariamente limitato to: upper GI serie, addominale ultrasound, cervello CT scansione, e esaminazioni per sinusitis, porphyria e gravidanza. Probably no singola individual richiede, o should receive, tutti del studi elencato, ed è importante to discuss the lavoro-up con your bambino’s medico. Questo è a molto nuovi e rapidamente evolvente field, e non nearly metà del answers sono in yet. Much of our understanding of, e hopefully our capacità per trattare, questo disturbo will migliora over the next numerosi mesi to anni. I am writing questo articolo a questo precoce stage in the speranza che alcune bambini sarà steered verso trattamenti ora le quali possono essere qualcosa aiuto per loro. Per maggiori informazioni, the Vomito coclico sindrome Association USA/Canada, an organization molto like la UMDF, può essere aiuto. I suggerire browsing loro web-site a: www.beaker.iupui.edu/cvsa http://www.beaker.iupui.edu/cvsa . In aggiunta, informazioni on ogni disponibili studi in CVS (con o senza malattia mitocondriale) e loro entrance criteri e procedure sono elencato there. Alternatively, you can reach the associazione by |
contacting: Debra Waites CVSA Administrator 3585 Cedar Hill Rd NW Canal Winchester, Ohio 43110 614-837-2586 cvsadwaites @msn.com http://www.beaker.iupui.edu/cvsa http://www.beaker.iupui.edu/cvsa> Riferimenti 1. Fleisher DR, The cyclic vomito sindrome descritte. J Pediatr Gastroenterol Nutr 1995; 21(Suppl. 1):S1-S5. 2. Li BUK, Vomito coclico: The modello e sindrome paradigmi. J Pediatr Gastroenterol Nutr 1995;21(Suppl. 1):S6-S10. 3. Boles RG, Chun N, Senadheera D, Wong L-JC (1997): Vomito coclico sindrome e DNA mitocondriale mutazioni. Lancet 350:1299-1300. 4. Boles RG, Williams JC. Mitocondriale malattia e Vomito coclico sindrome. Dig Dis Sci 1999;44(Suppl.):103S-107S. 5. Li, B U.K. e Balint, J. Vomito coclico sindrome, the evolution of understanding of a braingut disturbo. In Louis A. Barness (Ed.) Advances in pediatrica. St. Louis: Mosby Inc. 2000; 47,117-160. 6. Li, B U.K., Sarna, S . Issenman, R. (Guest Eds.). Proceedings del 2nd Scientifico simposio on CVS, Held a the Medical College of Wisconsin, USA, April 17-18, 1998. Digestive Malattie e Sciences. 1999;44(Suppl.):1S-120S. www.geneclinics.org Funded by Nazionale Institutes of Health (NIH) Health Resources e Services Adminstration (oreA) U.S. Department of Energy (DOE) Administrative Support: University of Washington School of Medicine bambini’s Hospital Regional Medical Center, Seattle, Washington |
Think
Mitocondri 63 UMDF
•
• !
• !
•
"
!
#
$
%
"
e
' (
)
*
$
+
, -
.
.
/001 233/
$
4 !
!"
! "
!" ! "
Come of June 2002, questo NIH-finanziato website è the la maggior parte delle comprehensive lista we’ve
trovati of esaminazioni facilities
per la malattia mitocondriale. Membership è libera. Log on e selezionati the “Laboratory
Directory” tab.
Benefici of UMDF Membership
Tri-annual Il bollettino
simposio Convegno Notices
Notices of Research Grant Opportunities (Goal of
$5.25 milioni di essere awarded in next 5 anni)
Educational Materials - per staff e pazienti
UMDF Patient Registry - A dinamica resource per
famiglie e specialisti facilitare lo scambio di informazioni (con scritto consent of I partecipanti)
Web Site - www.umdf.org - Access to Members
Solo Areas
Membership Application/Contribution Form
Enclosed sono my annual dues - entro the U.S.: $40
Enclosed sono my annual dues - outside del U.S.: $50 (drawn on U.S. bank)
Enclosed è my gift per aiutare sustain ricerche e famiglie supporto: $
(Please make checks payable to UMDF)
Per maggiori informazioni circa UMDF membership, please contatta la UMDF
office a 412-793-8077, info@umdf.org o visit www.umdf.org.
To Join ORA, please mail la forma sotto to:
The United Malattie mitocondriali Foundation
8085 Saltsburg Road, Suite 201
Pittsburgh, PA 15239
Please supply us con your informazioni to send a membership packet e/o to
acknowledge your gift:
Name: ____________________________ Address:
_____________________________________________________
City: ______________________________ State: ____________________________________
Zip: _______________
Office Phone: ______________________ Fax: _________________________ Email
__________________________
The sopra gift è composta: In Memory of _______________________ In Honor of
______________________
Name
_________________________________________________________________________________
On the occasion of: _____________________________________________ (Memorial,
birthdate, deathdate, altro)
Please send an acknowledgement of my gift to:
Name: ____________________________ Address:
_____________________________________________________
City: ______________________________ State: ____________________________________
Zip: _______________
UMDF anche accepts Visa o Mastercard
_______________________________________________________
(card numero)
Name come elencato on card (print): __________________________ Signature of
Cardholder: _________________________
Visit la UMDF Web Site per Upcoming Conferences a www.umdf.org
Mission Statement
To promote ricerche per cures e
trattamenti of Mitocondriale
Disorders e per fornire supporto
to persone affette e
famiglie.
Could it be Mitocondriale malattia?
ritardo nello sviluppo
visual e hearing problemi
acidosi lattica
malattia cardiaca
malattia epatica
attacchi epilettici
susceptibility to infezioni
debolezza muscolare
diabete
complicazioni respiratori
loss of motor control
gastro- intestinale malattie e deglutizione difficoltà
scarsa crescita
Think Mitocondri
Volume 1, June 2002 Questo pubblicazioni serve per aumentare awareness e informano
medica professionisti, famiglie
e persone circa
Mitocondriale malattia.
UMDF Mission
To promote ricerche per cures
e trattamenti of Mitocondriale
Disorders e per fornire supporto
to affetto famiglie.
UNITED MITOCHONDRIAL DISEASE FOUNDATION
8085 Saltsburg Road, Suite 201
Pittsburgh, PA 15239
Voice: 412-793-8077
Fax: 412-793-6477
Email: info@umdf.org
Web Site: www.umdf.org